EU MDR Transition Deadlines 2026, 2027 and 2028: The Complete Guide for Medical Device Manufacturers
Table of Contents
Introduction
EU MDR transition deadlines are now an immediate operational reality for every medical device manufacturer selling in the European Union. European medical device makers face a key May 26, 2026 deadline under the extended MDR transition timelines — the first cutoff date when Class III custom-made implantable devices must meet MDR requirements. For all other device classes, the clock is running toward December 31, 2027 and December 31, 2028.
This article provides the most complete and up-to-date guide to EU MDR transition deadlines available: exactly which deadlines apply to which device classes, what conditions must be met to benefit from the extended transition, what EUDAMED obligations are coming, and — most importantly — what manufacturers must do right now to protect their EU market access.
Background: Why the EU MDR Transition Was Extended
The EU Medical Device Regulation 2017/745 (EU MDR) became fully applicable on May 26, 2021, replacing the previous Medical Device Directive (MDD 93/42/EEC) and the Active Implantable Medical Devices Directive (AIMDD 90/385/EEC). From that date, all new devices placed on the EU market had to comply with the new regulation.
However, the original transition timeline quickly proved unworkable. With 21,376 products holding certificates issued under the old Directives and expiring by May 26, 2024, Notified Bodies estimated they could only issue around 7,000 MDR certificates by that date, creating the risk of massive device shortages across Europe.
In response, the European Commission adopted Regulation (EU) 2023/607 in March 2023. This amendment extended the MDR transition timelines and removed the earlier “sell-off” deadlines: Class III and most implantable Class IIb legacy devices can remain on the market until December 31, 2027, and other Class IIb, Class IIa, Class Is/Im, and certain up-classified Class I devices can remain until December 31, 2028, subject to strict conditions.
The removal of the sell-off period was a significant change — it means that qualifying legacy devices placed on the market before the transition deadline can continue to be sold without a fixed end date for stock clearance.
AUDIT-READY KIT
Build your ISO 13485 QMS with confidence.
Built on 15+ years of audit experience — every SOP and template references the regulations auditors expect. Get to certification faster, with industry best practices baked in.
- ✓30 SOPs covering the full QMS scope
- ✓56 templates ready to customize
- ✓Aligned with EU MDR + FDA QMSR
The Complete EU MDR Transition Deadline Timeline
The transition deadlines under Regulation 2023/607 are structured as follows:
- May 26, 2026: Class III custom-made implantable devices
- December 31, 2027: Class III and Class IIb implantable devices (except sutures, staples, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips and connectors)
- December 31, 2028: Class IIb (all other non-implantable), Class IIa, Class I sterile/measuring devices, and Class I devices up-classified under MDR

Figure 1 — EU MDR transition deadlines by device class
The May 26, 2026 Deadline — What Manufacturers Must Know Right Now
The May 26, 2026 deadline is the first hard deadline under the extended transition and applies exclusively to Class III custom-made implantable devices. This milestone marks a turning point for manufacturers that have been relying on legacy certificates under the old Medical Device Directive. By this date, any device that fails to meet the MDR requirements or obtain a new CE certificate could be pulled from the market.
Critically, custom-made Class III implantable devices do not benefit from the same conditions as other legacy devices — the MDR transition extension for custom-made Class III implantable devices does not specify any prerequisites: the extension is available even if the device was not MDD/AIMDD compliant before May 26, 2021 and even if the device is newly being placed on the market after the MDR’s date of application.
This means that manufacturers of custom-made Class III implantable devices cannot rely on the legacy device framework — they must achieve full MDR compliance by May 26, 2026, with no further extensions available.
What full MDR compliance means in practice:
- Complete technical documentation per Annex II and Annex III of EU MDR 2017/745
- Clinical evaluation report (CER) meeting the requirements of MDR Article 61 and MEDDEV 2.7/1 Rev 4
- Post-market surveillance (PMS) plan and system in place
- Quality management system (QMS) compliant with MDR Article 10(9) — typically demonstrated through ISO 13485 certification
- EUDAMED registration of the device and the manufacturer
- CE marking under MDR issued by a designated Notified Body
Conditions for Benefiting from the 2027 and 2028 Extended Deadlines
Not every manufacturer of legacy devices automatically benefits from the extended 2027 and 2028 deadlines. Regulation 2023/607 provides that these measures should not be applied indiscriminately but only to those companies that have already embarked on the path of MDR compliance.
To qualify, manufacturers must have met all three of the following conditions — all of which are now in the past:
Condition 1 — QMS implementation by May 26, 2024: Manufacturers had to implement a QMS in line with MDR Article 10(9) by May 26, 2024.
Condition 2 — Notified Body application by May 26, 2024: Manufacturers must have formally submitted an application to an MDR-designated Notified Body for conformity assessment of the legacy device by May 26, 2024.
Condition 3 — Written agreement with Notified Body by September 26, 2024: By September 26, 2024, manufacturers must have entered into a formal written agreement with the Notified Body for the conformity assessment.
If your company has not met these conditions, the extended transition deadlines do not apply to your devices. Your MDD/AIMDD legacy devices are no longer legally entitled to remain on the EU market under the transitional provisions, and you must achieve full MDR compliance immediately.
For devices with expired MDD certificates: Devices that had a valid MDD/AIMDD CE Marking certificate on May 26, 2021 but expired prior to March 20, 2023, can still qualify if the manufacturer either had a signed written agreement in place with the Notified Body or had been granted a derogation by an EU Competent Authority before the certificate expired.
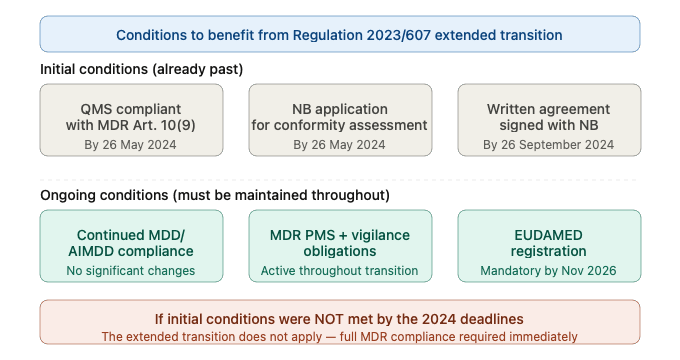
Figure 2 — Conditions for benefiting from EU MDR extended transition deadlines
The December 31, 2027 Deadline — High-Risk Devices
The December 31, 2027 deadline is the most consequential for the majority of high-risk medical device manufacturers. It applies to:
- All Class III devices (except custom-made implantable devices, which faced the May 2026 deadline)
- Class IIb implantable devices, with the following specific exceptions: sutures, staples, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips, and connectors — which fall under the 2028 deadline
Notified Bodies are reporting that the average time for a successful MDR certification review is 13 to 18 months, and for complex devices it can be longer. Waiting to submit an application puts manufacturers squarely in the bottleneck zone of 2026 and 2027, risking a lapse in the ability to sell the product in the EU.
This is the most critical practical implication of the 2027 deadline: if your conformity assessment is not already well advanced, you may not receive your MDR CE certificate before December 31, 2027, even if you submit your application today. The Notified Body review timeline alone — 13 to 18 months — means that applications should already be submitted or be submitted immediately for manufacturers targeting this deadline.
What must be complete and submitted to the Notified Body for Class III devices by the 2027 deadline:
Technical documentation (Annex II/III): Complete device description, design and manufacturing information, general safety and performance requirements (GSPRs) checklist, risk management file per ISO 14971, clinical evaluation report per Article 61 and Annex XIV, post-market surveillance plan, and labelling/IFU in all required EU languages.
Clinical evaluation: For Class III devices, the clinical evaluation requirements under EU MDR are significantly more demanding than under the MDD. The CER must demonstrate clinical evidence sufficient to support the device’s claims, and for many devices this will require clinical investigation data or a robust equivalence demonstration under MDCG 2020-5 criteria.
Quality management system: Full MDR-compliant QMS, typically ISO 13485 certified, covering design and development, manufacturing, post-market surveillance, CAPA, and all MDR-specific requirements including PRRC (Person Responsible for Regulatory Compliance) designation.
The December 31, 2028 Deadline — Lower-Risk Devices
The December 31, 2028 deadline applies to:
- Class IIb devices (non-implantable, and the specific implantable exceptions listed above)
- Class IIa devices
- Class I devices that are sterile, have a measuring function, or are reusable surgical instruments requiring Notified Body involvement
- Class I devices that were self-certified under MDD/AIMDD but are up-classified to a higher class under EU MDR
While the 2028 deadline provides more time than 2027, manufacturers should not be complacent. EUDAMED registration is mandatory — legacy devices must be registered by November 27, 2026. This means that even manufacturers targeting the 2028 transition deadline must complete EUDAMED registration well before their final compliance deadline.
The preparation timeline for Class IIa and IIb non-implantable devices is also substantial — typically 18 to 24 months from the start of technical documentation preparation to CE certificate issuance — meaning manufacturers who have not yet begun substantive MDR compliance activities are already behind schedule even for the 2028 deadline.
COMPLETE CATALOG
Find the documentation you need — instantly.
Whether you need a complete kit or just one specific SOP, our catalog has it. 45 process packages and 3 complete bundles, all instantly downloadable and fully editable.
- ✓Complete bundles or individual packages
- ✓45 process packages from €69 each
- ✓ISO 13485 · MDSAP · Combined Kit
EUDAMED — The Mandatory Registration Deadline
One of the most frequently overlooked aspects of the EU MDR transition is the EUDAMED (European Database on Medical Devices) registration obligation. Regulation 2024/1860 changes how EUDAMED goes live: instead of waiting until all modules are ready, the database will become mandatory module by module, with mandatory use beginning in phases from 2025 to 2026 for Actor registration, UDI/device registration, and Certificates/Notified Body data modules.
The key EUDAMED deadlines that manufacturers must be aware of:
Actor registration (manufacturer registration): Already mandatory. Every manufacturer selling medical devices in the EU must be registered as an Actor in EUDAMED before placing devices on the market.
UDI and device registration: Legacy devices must be registered in EUDAMED by November 27, 2026; this applies even to legacy devices still operating under the MDD/AIMDD transitional provisions.
Vigilance and post-market surveillance modules: Expected to become mandatory progressively through 2026 and 2027.
EUDAMED registration is not merely an administrative task — it requires manufacturers to assign Unique Device Identifiers (UDIs) to their devices, register all device models and variants, maintain accurate and up-to-date product information, and link their registration to their Notified Body certificates. For manufacturers with large product portfolios, EUDAMED registration can be a substantial project requiring months of data preparation.
The Notified Body Bottleneck — The Most Urgent Risk
The final MDR transition phase will test more than regulatory compliance — it will test operational resilience across the entire medical device industry. 2026 and 2027 will be the most demanding years yet, and those who delay may face certification gaps, market disruption, or withdrawal of devices from the EU market.
The Notified Body capacity constraint is not theoretical — it is already affecting manufacturers. The number of MDR-designated Notified Bodies remains limited, their review queues are growing, and the complexity of MDR technical documentation means review cycles are substantially longer than under the MDD.
Data presented by Team NB indicates that Notified Body reviews of technical documentation under MDR 2017/745 are mostly taking up to 18 months. That does not necessarily mean that the review can safely take place in 2026 or 2027 in order to be complete before the 2027 or 2028 deadlines, respectively.
The practical implication is stark: for the 2027 deadline, manufacturers who are not already deep into their conformity assessment process are at serious risk of missing the deadline, regardless of how well-prepared their technical documentation is. Notified Body slots are finite, and the industry-wide rush toward the 2027 and 2028 deadlines is creating a bottleneck that will only worsen as the deadlines approach.
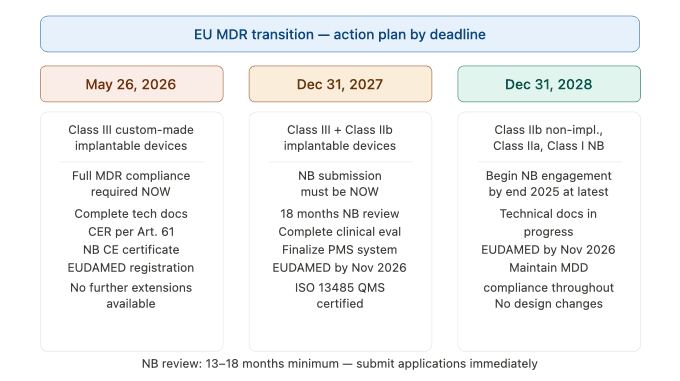
Figure 3 — EU MDR transition action plan by deadline
How to Demonstrate Legacy Device Status
One practical challenge that many manufacturers face is how to demonstrate to customers, distributors, and market surveillance authorities that their legacy devices remain legally on the market despite apparently expired MDD certificates. There will be no new certificates with new expiry dates issued by Notified Bodies under the MDD/AIMDD — the extension of a certificate’s validity is done automatically by law as long as the conditions for extension are fulfilled.
The European Commission and industry associations have outlined several approaches:
Manufacturer self-declaration: Manufacturers can generate a self-declaration statement confirming that the extension requirements per Regulation 2023/607 have been fulfilled. The statement should clearly identify the devices covered by the extension and the CE certificate(s) to which it applies. MedTech Europe has published a template for this purpose.
Notified Body confirmation letter: The Notified Body could issue a confirmation letter stating the receipt of the manufacturer’s application for conformity assessment and the conclusion of a written agreement, clearly identifying the devices covered by the extension and applicable CE certificates.
Certificates of Free Sale: Manufacturers or their authorized representatives can request Certificates of Free Sale from Competent Authorities, which can be issued noting the extended CE marking validity period.
Any of these documents, when provided to customers or customs authorities, serves as evidence that the device remains legally on the EU market despite an expired certificate date.
What Happens If You Miss the Deadline?
The consequences of failing to achieve MDR compliance by the applicable transition deadline are severe and immediate. Devices that do not have a valid MDR CE certificate by their deadline — and do not qualify for the transitional provisions — must be withdrawn from the EU market. This means they cannot be placed on the market, sold to distributors, or transferred to end users.
Devices that are already on the market (already placed) may continue to be sold and used by end customers — this is the practical effect of the removal of the sell-off period by Regulation 2023/607. However, no new stock can be placed on the EU market after the deadline. For manufacturers with complex supply chains, this distinction between “placed on the market” and “in use” requires careful legal analysis.
The financial consequences of a market withdrawal can be devastating — particularly for manufacturers who rely on the EU market for a significant portion of their revenue. Beyond the immediate revenue impact, the reputational damage, the costs of emergency regulatory remediation, and the potential impact on patients who depend on the device all make late compliance extremely costly.
Practical Action Plan for Manufacturers — What to Do Now
Regardless of which deadline applies to your devices, the actions required are fundamentally the same — the only variable is urgency.
Immediately — assess your portfolio status: Map every product in your portfolio against the transition deadlines. For each device, determine whether it qualifies as a legacy device under Regulation 2023/607, which deadline applies, and whether the 2024 qualification conditions were met. This portfolio analysis is the essential starting point for any MDR transition strategy.
Immediately — engage your Notified Body: If manufacturers haven’t initiated discussions with their Notified Body, they should start immediately, as slots will become scarce as deadlines approach. Even if your technical documentation is not yet complete, early engagement with your Notified Body allows you to understand their specific requirements, timeline expectations, and documentation preferences — all of which will make the formal review faster and smoother.
Within 3 months — complete your technical documentation gap analysis: Compare your existing MDD technical file against the EU MDR Annex II and III requirements. Identify the gaps — typically the most significant gaps are in clinical evaluation depth, post-market surveillance, and EUDAMED-related documentation.
Within 6 months — complete EUDAMED registration: Even if your transition deadline is 2027 or 2028, EUDAMED registration for legacy devices is required by November 2026. Begin UDI assignment and device registration immediately to avoid this becoming a last-minute bottleneck.
Ongoing — maintain full MDD/AIMDD compliance: Until MDR certification is achieved, the legacy device must remain fully compliant with the original directive under which it was certified. Any significant design change risks invalidating the transition extension.

Figure 4 — EU MDR transition risk assessment matrix
Frequently Asked Questions on EU MDR Transition Deadlines
Can I still sell my MDD-certified device in the EU after the transition deadline? No — once the transition deadline for your device class has passed, you cannot place new stock on the EU market without a valid MDR CE certificate. Stock that was already placed on the market (i.e., transferred to distributors or end users) before the deadline can continue to be sold and used, but new placements require MDR certification.
Do I need a new Notified Body for my MDR certification? Not necessarily — if your current MDD Notified Body is also designated under EU MDR, you can continue working with them. However, if your Notified Body is not MDR-designated, you must engage a new one before May 26, 2024. Check the NANDO database for current MDR-designated Notified Bodies.
My MDD certificate expired before March 20, 2023 — can I still benefit from the transition? Potentially yes, if you had a signed written agreement with a Notified Body for MDR conformity assessment before the certificate expired, or if you obtained an Article 59(1) derogation or Article 97(1) authorization before expiry. The specific conditions are complex and you should seek specialist regulatory advice.
What counts as a “significant change” that would invalidate my legacy device status? MDCG 2020-3 provides guidance on what constitutes a significant change. Generally, changes that affect the device’s safety, performance, or intended purpose — changes to design, materials, manufacturing processes, or sterilization methods — are likely to be considered significant. Minor changes to labelling or documentation are generally not significant. Consult the MDCG guidance and, where uncertain, seek Notified Body confirmation before making any changes.
Is EUDAMED registration mandatory for legacy devices? Yes — even legacy devices operating under the transitional provisions must be registered in EUDAMED. The mandatory registration deadline for legacy devices is November 27, 2026, regardless of whether the device’s transition deadline is 2027 or 2028.
Conclusions
The EU MDR transition deadlines of 2026, 2027, and 2028 represent the final chapter of one of the most complex regulatory transitions in the history of the medical device industry. The extended timeline provided by Regulation 2023/607 was a necessary response to the realities of Notified Body capacity and industry readiness — but the extensions have an end, and that end is now approaching rapidly.
For manufacturers of Class III custom-made implantable devices, May 26, 2026 is an immediate and absolute deadline with no further extensions available. For manufacturers of Class III and Class IIb implantable devices, December 31, 2027 is less than two years away — and given Notified Body review timelines of 13 to 18 months, the window for action is effectively now. For all other device classes, the 2028 deadline provides marginally more time — but EUDAMED registration obligations in 2026 mean that no manufacturer can defer MDR compliance activities much further.
The manufacturers who will successfully navigate these deadlines are those who treat MDR compliance not as a regulatory burden but as a business-critical project — one that requires dedicated resources, proactive Notified Body engagement, and systematic technical documentation development starting today.
This article is part of the MD Regulatory series on EU MDR compliance. Related articles cover EU MDR technical documentation requirements, clinical evaluation under EU MDR, EUDAMED registration, and post-market surveillance obligations.





3 Comments
Comments are closed.