The Benefit-Risk Analysis: Avoid Critical Mistakes and Achieve Full Compliance
Table of Contents
Benefit-risk analysis represents a fundamental component of the risk management process for medical devices and, more broadly, a cornerstone of any effective quality management system. It serves as a critical mechanism for evaluating whether the overall advantages of a device outweigh its potential risks, thereby supporting informed decision-making throughout the product lifecycle.
The topic of risk management has been extensively addressed, with particular reference to the requirements outlined in ISO 14971:2019 and the supporting guidance provided in ISO/TR 24971:2020. In addition, risk management principles have been explored across other life science sectors, including the pharmaceutical industry, where frameworks such as the ICH Q9 guideline establish a systematic approach to quality risk management.
The benefit-risk analysis is an essential methodology nowadays to be used to complete documentation for MDR or 510k certification or related market-access certifications (MDSAP).
In this article we will deal in details with different methologies to be used for the determination of the benefit-risk analysis, with a particular focus on EU MDR and the connection between risk management and clinical-related documentation, such as clinical evaluation plan and report.
MULTI-MARKET KIT
One audit. Five markets. Ready to submit.
Our MDSAP Documentation Kit covers Brazil ANVISA, Japan PMDA, Canada Health Canada, Australia TGA, and FDA — with country-specific reportability worksheets and application checklists you can use today.
- ✓15 SOPs covering 5 MDSAP markets
- ✓18 templates with country worksheets
- ✓Brazil · Japan · Canada · Australia · USA
Benefit-Risk Analysis under EU MDR 2017/745: Key Requirements and Practical Implications
With the entry into force of the EU Medical Device Regulation (EU MDR 2017/745), the expectations surrounding risk management for medical devices have significantly increased, particularly in relation to benefit-risk analysis. This is no longer a standalone exercise but a central, continuously evolving process that must be fully integrated within the broader quality management system (QMS) and benefit-risk analysis plays a pivotal role in the context.
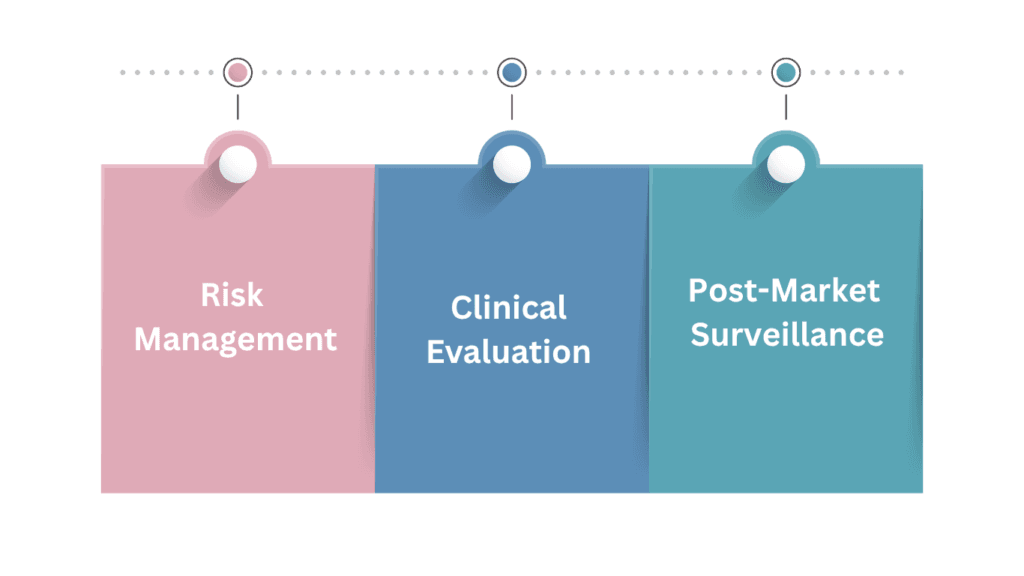
One of the most critical shifts introduced by the MDR is the explicit interconnection between risk management, clinical evaluation, and post-market surveillance (PMS). These three elements must function as a cohesive system, ensuring that the device remains safe and effective throughout its entire lifecycle—not only at the time of market entry but also during real-world use.
In this article, we explore in detail the regulatory expectations for benefit-risk analysis under EU MDR 2017/745, providing clarity on definitions, documentation requirements, and lifecycle integration.
Understanding Benefit-Risk Analysis
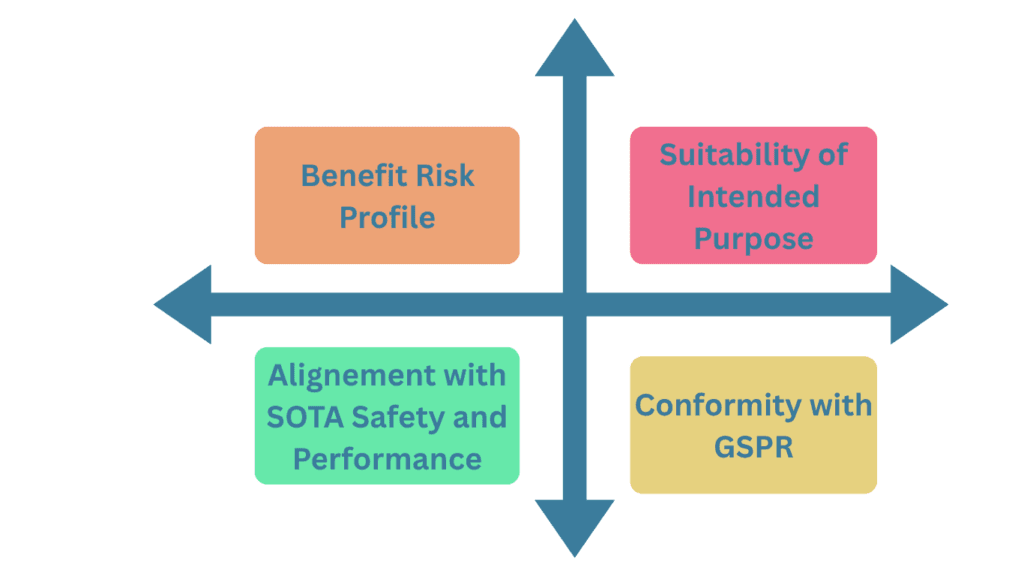
A solid starting point is the regulatory definition. According to Article 2 of the EU MDR, benefit-risk analysis (or determination) refers to the comprehensive analysis of all relevant benefits and risks associated with the use of a medical device for its intended purpose, as defined by the manufacturer.
This definition highlights a crucial concept: the benefit-risk analysis must be context-specific, meaning that risks and benefits are not assessed in isolation but always in relation to the intended use, target population, and conditions of use.
Closely linked to this is the concept of clinical benefit, which plays a pivotal role in any benefit-risk analysis. Under the MDR, clinical benefit is defined as the positive impact of a device on patient health, expressed through measurable, clinically meaningful outcomes. These outcomes may relate to diagnosis, therapeutic effect, patient management, or even broader public health improvements.
Understanding and clearly demonstrating clinical benefit is essential, as it forms the foundation upon which the acceptability of risks is justified and the construction of a solid benefit-risk analysis.
Benefit-Risk Analysis within Technical Documentation
The EU MDR places strong emphasis on documenting the benefit-risk profile within the technical documentation. As specified in Annex II, manufacturers are required to provide a clear benefit-risk analysis, demonstrating that all identified risks are acceptable when weighed against the expected clinical benefits. In addition, objective evidence must be included to support the establishment and implementation of a comprehensive risk management system, ensuring that the benefit-risk ratio is systematically evaluated, justified, and documented.
A major innovation introduced by the MDR is the reinforcement of post-market surveillance as a driver for continuous benefit-risk evaluation. According to Annex III, the PMS plan must define relevant indicators and predefined threshold values that are used to continuously reassess the benefit-risk profile once the device is placed on the market.
This requirement ensures that manufacturers actively monitor real-world performance and promptly identify any changes that could affect safety or effectiveness.
The importance of lifecycle thinking is further emphasized in Annex XIV, Part B, which requires that the Post-Market Clinical Follow-Up (PMCF) plan clearly describes how the continued acceptability of the benefit-risk ratio will be evaluated. In practice, this involves ongoing clinical data collection, confirmation of sustained clinical benefits, and reassessment of risks based on newly available evidence.
Under EU MDR 2017/745, benefit-risk analysis is no longer a static regulatory requirement but a continuous, data-driven process embedded across risk management, clinical evaluation, and post-market activities. Manufacturers are expected to adopt a proactive and integrated approach, ensuring that the benefit-risk analysis remains valid, justified, and supported throughout the entire lifecycle of the device.
This evolution marks a shift from a purely compliance-driven model to one focused on continuous assurance of safety and performance, ultimately strengthening patient protection and product quality.
Step‑by‑Step Guide: How to Perform Benefit‑Risk Analysis under EU MDR
Conducting a benefit‑risk analysis under the European Medical Device Regulation (EU MDR 2017/745) is a complex, continuous, and evidence‑based process that extends across the product lifecycle. Its purpose is to demonstrate that the benefits of a device outweigh its risks in accordance with regulatory expectations, specifically as defined in Article 2 of the MDR and reinforced in general safety and performance requirements (GSPRs).
Unlike simple risk assessments, benefit‑risk analysis must move beyond listing hazards and controls — it must systematically compare benefits and risks, using clinical data, state‑of‑the‑art references, and a structured evaluation framework to justify risk acceptability. Here’s how to do it step by step.
Define the Scope and Intended Use
The entire benefit–risk analysis must be grounded in the precise regulatory definition of the device’s intended use, as both benefits and risks can only be meaningfully evaluated within this context. In practical terms, this requires first confirming the intended purpose and indications as specified by the manufacturer, then clearly identifying the target patient population, the clinical or procedural setting in which the device is used, and the profile of the intended users. It is also essential to determine whether the device is intended to support diagnosis, treatment, monitoring, or other clinical outcomes. Establishing this framework is critical, as it directly guides the selection and interpretation of clinical evidence, as well as the identification of relevant risks and the definition of measurable benefit parameters.
Conduct a Thorough State‑of‑the‑Art Analysis
Before any meaningful comparison can be undertaken, the manufacturer must establish a clear and well-documented understanding of the state of the art (SotA) for comparable devices and available therapeutic alternatives. This step is essential, as the SotA provides a scientifically grounded benchmark for evaluating both expected benefits and acceptable levels of risk based on existing solutions. It also enables the identification of relevant clinical alternatives—such as pharmacological treatments, surgical interventions, or competing medical devices—against which the device can be assessed. In addition, defining the SotA helps map the broader medical landscape, including how clinical outcomes are measured and what constitutes meaningful performance in real-world practice. Regulatory authorities and Notified Bodies require that the SotA be established independently, supported by robust references, and fully integrated into both the clinical evaluation plan and the clinical evaluation report. Without a comprehensive and objective SotA, it becomes extremely challenging to justify a benefit–risk analysis that is scientifically credible and compliant with regulatory expectations.
Identify and Catalogue Relevant Benefits
Begin by defining what constitutes a benefit in the context of the device. Under the MDR, clinical benefits include measurable, patient‑relevant outcomes, such as improved diagnosis, therapeutic outcomes, reduced morbidity or mortality, or enhanced patient management.
Benefits should be categorized, for example:
- Direct clinical benefits — e.g., improved survival, symptom relief.
- Indirect benefits — e.g., reduced procedure time, decreased hospital stays.
- Broader impacts — e.g., improved quality of life, reduced caregiver burden.
Each identified benefit must be supported by clinical evidence, whether from literature, clinical investigations, registries, or real‑world data.
Identify and Characterize Risks with Clinical Context
Simultaneously, all relevant risks must be identified and contextualized with clinical evidence. This goes beyond technical risk management (ISO 14971) and requires clinical insight into how risks manifest in the real world.
Consider:
- Device‑specific risks — e.g., material reactions, device failure modes.
- Procedure‑related risks — e.g., surgical complications or algorithm misinterpretation.
- User‑related risks — e.g., misuse, training deficiencies.
For each risk, characterize:
- Severity: how serious the outcome could be.
- Probability: how frequently it might occur.
- Clinical relevance: impact on patient management or health outcomes.
This step should integrate risk data from ISO 14971 files and clinical sources, ensuring a comprehensive risk catalog.
Establish Quantitative and Qualitative Parameters for Comparison
Relying exclusively on qualitative descriptions can result in conclusions that are subjective and open to interpretation; therefore, once benefits and risks have been systematically identified, it is essential to define structured evaluation parameters that enable a balanced, transparent, and reproducible comparison. These parameters should incorporate elements such as the frequency of occurrence—comparing the incidence of clinical benefits against that of adverse events—the magnitude of effect, assessing the clinical relevance of the benefit in relation to the severity of associated risks, and the duration, considering how long the benefit persists relative to the period of risk exposure. In addition, it is important to evaluate the impact across different patient populations, taking into account variability among subgroups and demographic factors. Whenever feasible, outcomes should be supported by quantitative methods, including numerical indicators such as the number needed to treat (NNT) or number needed to harm (NNH), as well as comparative metrics that benchmark performance against alternative treatments or existing standards of care. For aspects that cannot be readily quantified, well-justified qualitative descriptors should still be incorporated. This structured and multi-dimensional approach is critical for transforming raw clinical data into a coherent, evidence-based argument that supports regulatory justification of the benefit-risk analysis .
✦ PREMIUM BUNDLE
The ultimate global QMS documentation bundle.
Combine ISO 13485 + all 5 MDSAP markets in one premium package. Deduplicated structure means you customize each document once — not twice. Save €199 vs buying the kits separately.
- ✓41 SOPs covering both ISO 13485 and MDSAP
- ✓70+ templates with deduplicated structure
- ✓Save €199 vs buying separately
Perform the Comparative Analysis
With benefits and risks outlined and comparable parameters defined, the core analysis begins: weighing benefits against risks. This is not a single calculation but an integrated narrative that answers:
- Do the benefits meaningfully outweigh the risks?
- Are the observed risks acceptable in the context of the patient benefit?
- How does the device compare to alternatives under SotA considerations?
- Are there specific subgroups where the balance differs?
This step synthesizes evidence from systematic literature, clinical investigations, PMS data, and SotA benchmarks. Quantitative and qualitative insights should be woven together, recognizing that not all outcomes can be reduced to a single score
Draft a Clear, Evidence‑Based Benefit‑Risk Justification
Once the comparative assessment has been completed, its outcomes must be translated into a coherent and well-structured narrative suitable for integration into key regulatory documents required under the MDR, including the Clinical Evaluation Report (CER), the Risk Management File (RMF), and the Post-Market Surveillance (PMS) documentation. This narrative should not merely summarize findings, but rather present a clear, evidence-based justification of the benefit-risk analysis, ensuring consistency across all elements of the technical documentation. A robust benefit–risk analysis is expected to explicitly reference the underlying clinical evidence and the established state of the art, synthesize the most relevant benefits alongside the contextualized risks, and demonstrate how applicable regulatory requirements—such as the General Safety and Performance Requirements (GSPR), particularly points 1 and 8—are fulfilled. It should also transparently address any residual uncertainties, data gaps, or methodological limitations, providing a balanced interpretation of the available evidence. Ultimately, the narrative must culminate in a reasoned and defensible conclusion on the overall acceptability of the benefit–risk ratio, aligned with both regulatory expectations and clinical relevance.
Integrate PMS and PMCF Data for Ongoing Reevaluation
The EU MDR clearly establishes that benefit–risk analysis is not a one-off regulatory activity, but a dynamic and continuous process that must be maintained throughout the entire lifecycle of the device. Post-market data play a central role in this approach, as they are expected to systematically feed back into the ongoing reassessment of the benefit–risk profile. In practical terms, this requires the implementation of robust PMS indicators and predefined threshold criteria to detect emerging safety signals and performance trends. As new evidence becomes available, the benefit-risk analysis must be proactively reviewed and, where necessary, updated to reflect the latest data. This includes the structured integration of findings from Post-Market Clinical Follow-Up (PMCF) activities into periodic updates of the Clinical Evaluation Report. Moreover, changes observed in real-world device performance or shifts in the state of the art may alter the balance between benefits and risks over time, potentially triggering the need for adjustments in labeling, risk control measures, or even clinical claims. Maintaining this continuous feedback loop is essential to ensure sustained compliance, patient safety, and clinical relevance under the MDR framework.
Conclusions
Benefit–risk analysis under the EU MDR is no longer a static or purely formal requirement, but a central, evidence-driven process that underpins the entire lifecycle of a medical device. From the initial definition of intended use to the establishment of the state of the art, from the structured evaluation of clinical benefits and risks to their continuous reassessment through PMS and PMCF activities, every step contributes to building a robust and defensible justification of the device’s safety and performance.
A well-executed benefit–risk analysis goes beyond compliance: it demonstrates scientific rigor, clinical relevance, and a deep understanding of how the device performs in real-world conditions. It requires the integration of multiple data sources, the use of both quantitative and qualitative methodologies, and a transparent acknowledgment of uncertainties and limitations.
Ultimately, the strength of the benefit–risk determination lies in its ability to clearly show that the device delivers meaningful clinical value while maintaining risks at an acceptable level in light of the current state of the art. In an increasingly stringent regulatory environment, this is not only essential for achieving and maintaining CE marking, but also for ensuring long-term trust from regulators, healthcare professionals, and patients.