EUDAMED Registration Requirements: Step-by-Step Guide for Medical Device Manufacturers
Table of Contents
- Introduction
- Why EUDAMED Registration Is Now Mandatory
- The Key 2026 Deadlines You Cannot Miss
- Who Must Register — A Decision Path
- The Six EUDAMED Modules
- Step-by-Step: How to Register in EUDAMED
- Machine-to-Machine Integration: When and Why
- Common Registration Mistakes and How to Avoid Them
- Practical Tips for a Tight Timeline
- Frequently Asked Questions
- Conclusions
Introduction
Understanding the EUDAMED registration requirements for medical devices is now urgent rather than optional. On 27 November 2025 the European Commission published Commission Decision (EU) 2025/2371, formally confirming that four EUDAMED modules are fully functional and starting a six-month countdown. From 28 May 2026, registration in EUDAMED is mandatory: without a valid Single Registration Number (SRN), an economic operator can no longer place devices on the EU market, and parallel national systems no longer suffice. This guide is a step-by-step walkthrough of EUDAMED registration — actor registration, UDI and device registration, the mandatory 2026 deadlines, the common mistakes that cause rejection, and practical tips for manufacturers preparing under a tight timeline.
It is written for regulatory and quality professionals who need to act now, and it connects to the wider compliance picture in our EU MDR transition deadlines, EU MDR technical documentation and UDI requirements guides. Throughout, the emphasis is on what to do in practice rather than on restating the regulation: with the timeline now fixed, execution is the only thing that matters.
Why EUDAMED Registration Is Now Mandatory
EUDAMED is the European Database on Medical Devices — the central IT system through which the Commission, competent authorities, manufacturers and Notified Bodies share regulatory information under the EU MDR (Regulation (EU) 2017/745) and IVDR (Regulation (EU) 2017/746). For years it was available only for voluntary use, and its deadlines were repeatedly postponed, which led many organisations to treat it as a problem for the future. That has now changed decisively. Under Regulation (EU) 2024/1860, EUDAMED is deployed module by module: each module becomes legally mandatory six months after the Commission formally declares it functional in the Official Journal. Decision (EU) 2025/2371 was that formal notice for the first four modules, and it converted a long-running “someday” obligation into a hard, dated one.
The practical consequence is stark. From 28 May 2026, any regulatory obligation linked to one of the four mandatory modules must be fulfilled in EUDAMED. Actor registration becomes a gatekeeper: without an SRN you cannot complete device registration or place devices on the market. User-interface difficulties or operational excuses will no longer justify non-compliance — the Commission has been explicit that the validated functionality of the four modules removes the prior justification for delay. For organisations that have not begun, the preparation window is measured in months, and a great deal of that time is consumed by data preparation and validation rather than by the act of submission itself.
There is also a strategic dimension that is easy to miss. EUDAMED is not merely a registration formality; it becomes the authoritative public and regulatory data source for devices on the EU market. Competent authorities will use it to coordinate market surveillance, Notified Bodies will use it for certificate management, and any inconsistency between EUDAMED and a manufacturer’s other documentation becomes visible and actionable. This means the registration project is also a data-governance project: the goal is not just to get records into the system before the deadline, but to get them in correctly, because incorrect records now carry regulatory and legal consequences rather than being a private housekeeping matter.
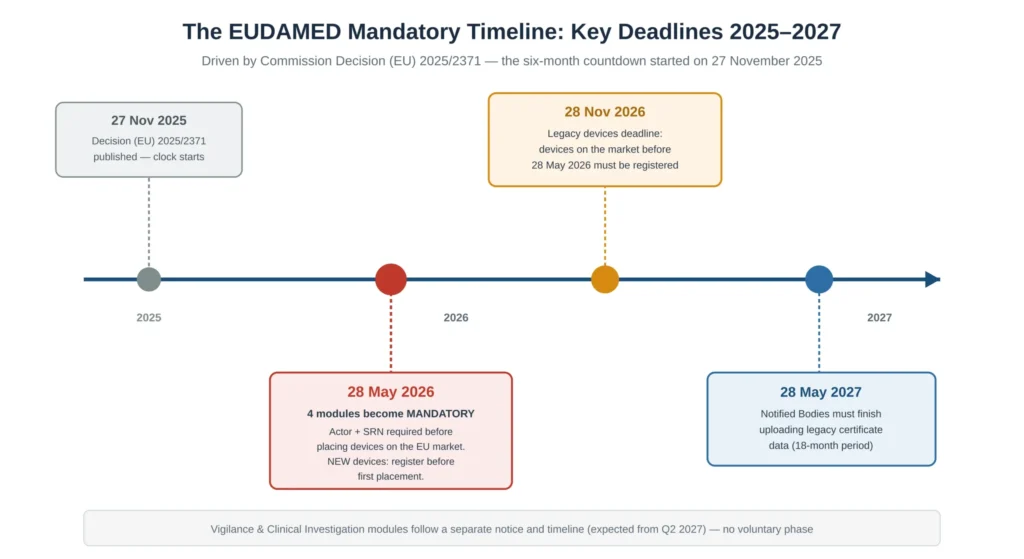
The Key 2026 Deadlines You Cannot Miss
Three dates anchor every EUDAMED compliance plan, and confusing them is one of the most common planning errors.
28 May 2026 is the headline date. The four modules become mandatory, and from that day a new MDR or IVDR device cannot be placed on the EU market unless its device record exists in EUDAMED and the responsible operator holds a valid SRN. This is an absolute precondition: it is not sufficient to have started the process, the records must actually be in place before first placement.
28 November 2026 is the legacy-device deadline. Devices that were placed on the market before 28 May 2026 and continue to be marketed afterwards must be registered by this date — twelve months after the notice took effect. This deadline is frequently underestimated because legacy portfolios are often large, the data is often scattered across legacy systems, and the work of creating Basic UDI-DIs retrospectively for older products is slow.
28 May 2027 is the Notified Body certificate deadline. Notified Bodies must complete the upload of relevant legacy certificate information within the 18-month period. While this is the Notified Body’s obligation rather than the manufacturer’s, manufacturers are affected: during the interim, manufacturers should maintain accessible copies of certificates for distributors and importers, and should confirm the upload schedule with their Notified Body rather than assuming it will be handled silently.
Two further points are commonly misunderstood. First, EUDAMED registration runs on its own schedule, independent of the MDR/IVDR transition periods for certification — meeting an MDR certificate deadline does not exempt you from the EUDAMED dates, and the two timelines must be planned as separate workstreams that happen to interact. Second, legacy and MDR devices that are no longer placed on the market from 28 May 2026 do not generally need to be registered, unless a post-market surveillance or vigilance action occurs for them; this is a useful scoping relief for discontinued product lines, but it must be documented deliberately rather than assumed.
✦ EU MDR TECHNICAL DOCUMENTATION KIT
The technical file EUDAMED will be checked against.
The complete Annex II technical file in one coordinated set: Device Description and Specifications, Labelling and Instructions for Use, Design and Manufacturing Information, General Safety and Performance Requirements, Benefit-Risk Analysis and Risk Management, Verification and Validation, Post-Market Surveillance and the Declaration of Conformity — the controlled source your EUDAMED device records must match.
- ✓ 8 Word templates · full Annex II coverage · Annex III and Annex IV included
- ✓ MDCG guidance and Team-NB position papers integrated
One-time €429
Get the MDR Tech Doc Kit →Who Must Register — A Decision Path
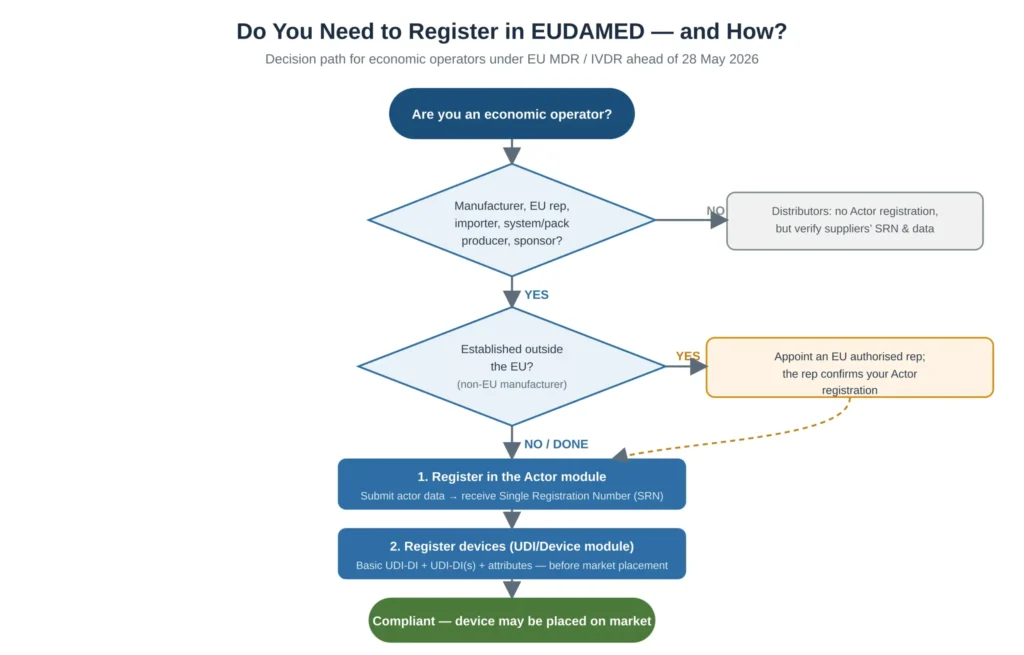
Not every organisation in the supply chain has the same obligation, and applying the wrong assumption is a costly error this close to the deadline.
All economic operators in scope of Article 31 MDR / Article 28 IVDR must complete Actor registration and hold an SRN before placing devices on the market. This includes manufacturers, EU authorised representatives, importers, system and procedure pack producers, and sponsors of clinical investigations and performance studies. Each of these roles has its own data requirements and its own obligations once registered, so the first analytical task is to enumerate every legal entity in your corporate structure that performs one of these functions — a single group may contain several distinct actors that each need their own registration.
Distributors are not economic operators that complete Actor registration in the same way as the operators above. However, distributors are not exempt from EUDAMED-related diligence: they carry a practical obligation to verify that their suppliers are registered, that the devices they handle have device records, and that the data is consistent. A distributor that continues to place a device on the market when the upstream operator has failed to register it is exposed, so distributor verification procedures should be updated to include an EUDAMED check.
Non-EU manufacturers face an additional step that is a frequent bottleneck. They must register in the Actor module and must also have an EU authorised representative who confirms the registration. The dependency runs in a specific order: the representative relationship must be in place for the confirmation to occur, so a non-EU manufacturer that has not yet appointed or formalised its EU authorised representative cannot complete Actor registration regardless of how quickly it prepares its own data. This should be the very first item on a non-EU manufacturer’s plan.
For custom-made devices a narrower rule applies. Broadly, only manufacturers of custom-made class III implantable devices must register as an actor so that the Notified Body can register the corresponding QMS certificate; manufacturers of only custom-made devices generally must register in the Actor module before they submit vigilance or post-market surveillance information, but the device-registration burden differs from that of series-manufactured devices. Because the custom-made rules are nuanced, manufacturers in this category should confirm their specific obligation rather than applying the general rule by analogy.
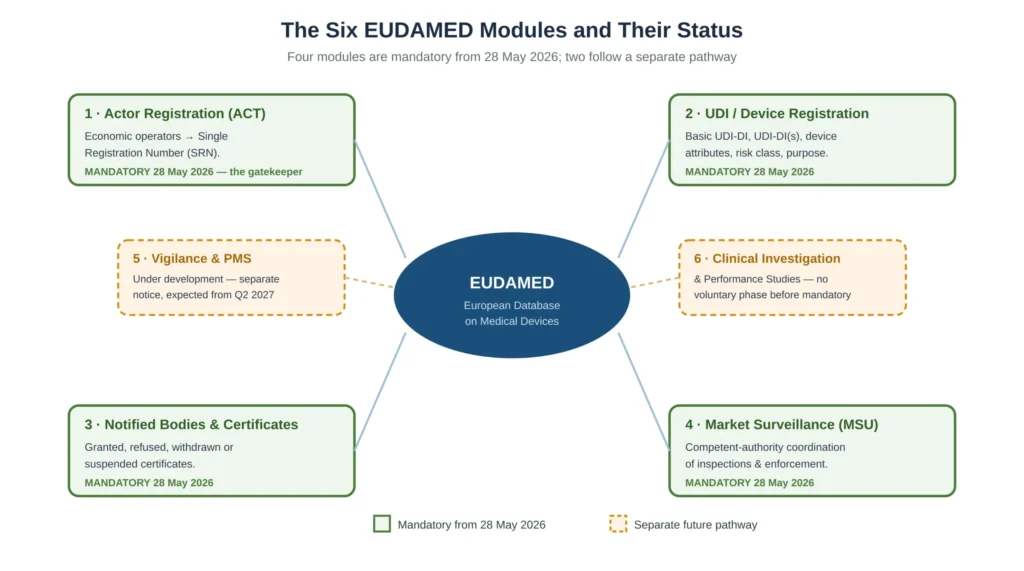
The Six EUDAMED Modules
EUDAMED is built from six modules, and knowing which are in force shapes what you must do now.
Four are mandatory from 28 May 2026. Actor Registration is the entry point that issues the SRN and gates everything else. UDI/Device Registration holds device identification, the Basic UDI-DI, the UDI-DIs and the device attributes such as risk class and intended purpose. Notified Bodies and Certificates holds certificate information, including granted, refused, withdrawn and suspended certificates — and notably, refused applications become visible across Notified Bodies, which means a manufacturer’s prior failures are no longer private. Market Surveillance is used by competent authorities to coordinate inspections and enforcement, and a market-surveillance report filed by any one Member State becomes instantly visible across the EU.
The remaining two modules — Vigilance and Post-Market Surveillance, and Clinical Investigation and Performance Studies — are still under development and follow a separate notice and timeline, expected from Q2 2027. A specific point matters here: unlike the first four modules, these two will have no voluntary-use phase before they become mandatory. There will be no period of optional familiarisation, so manufacturers should not assume they will get a soft introduction to these modules the way the first four had years of voluntary availability.
The visibility characteristics of the certificate and market-surveillance modules deserve emphasis because they change behaviour. When refused certificates and enforcement actions become EU-wide visible, the cost of inconsistent or inaccurate data rises sharply: an error is no longer a local administrative issue but a publicly and cross-authority visible discrepancy. This is another reason the registration project must be treated as a data-quality exercise rather than a box-ticking one.
Step-by-Step: How to Register in EUDAMED
The registration process is sequential, and each step is a hard precondition for the next. There is no shortcut around the order: no SRN means no device registration, and no device record means no market placement. Figure 4 lays out the practical sequence; the narrative below explains each step.
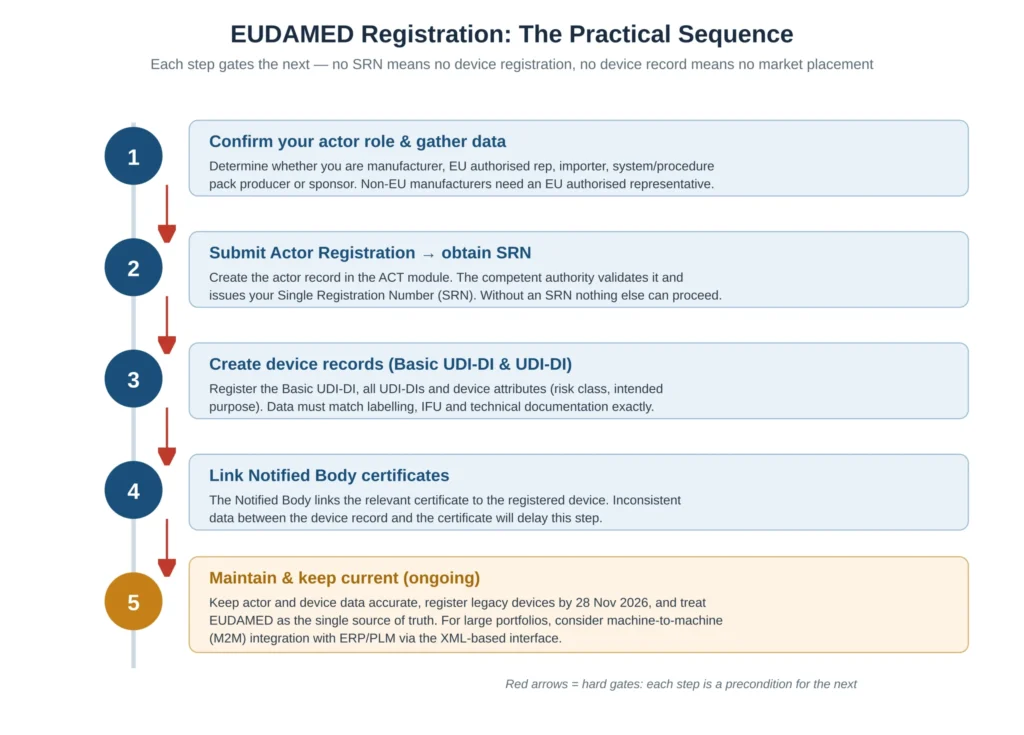
Step 1: Confirm Your Actor Role and Gather Data
Before touching the system, determine precisely which actor role applies to your organisation — manufacturer, EU authorised representative, importer, system or procedure pack producer, or sponsor — and do this for every legal entity in your group, not just the headline manufacturer. The role determines what data you submit and what obligations follow, and getting it wrong propagates errors into every subsequent step.
Non-EU manufacturers must establish an EU authorised representative now, because the representative confirms the Actor registration and a missing or unprepared representative is the single most common bottleneck for non-EU organisations. Gather the legal entity details, contact information and supporting documentation in advance, because incomplete actor data delays competent-authority validation, and that validation time is outside your control once you submit. The practical tip here is to prepare the actor dossier as if it were a regulatory submission in its own right: complete, internally consistent, and reviewed before it goes in.
Step 2: Submit Actor Registration and Obtain the SRN
Create the actor record in the Actor (ACT) module. The relevant competent authority reviews and validates the submission and then issues the Single Registration Number. This validation is not instantaneous, which is precisely why starting early matters: the SRN is the key that unlocks every other function in EUDAMED, and you cannot register a single device without it. Treat the SRN as a critical-path item in your 28 May 2026 plan, not an administrative afterthought, and build in contingency time for the possibility that the competent authority returns the submission for correction. An organisation that submits its actor data in, say, April 2026 and is asked for clarification has very little margin; one that submits months earlier has a comfortable buffer.
Step 3: Create Device Records
With an SRN in hand, register devices in the UDI/Device module: the Basic UDI-DI, all relevant UDI-DIs, and the device attributes including risk class and intended purpose. The single most important practical tip is consistency. The EUDAMED data must match the device labelling, the instructions for use, the technical documentation and the Notified Body certificate exactly. “Exactly” is not rhetorical: differences in product naming, intended-purpose wording, or risk class between these sources are a leading cause of delay and can hold up certification and surveillance actions.
A useful way to operationalise this is to designate a single controlled source for each data element before any record is created — for example, deciding that the intended-purpose statement in the approved technical documentation is the master text, and that the EUDAMED entry, the label and the IFU must all derive from it without paraphrase. Doing this mapping work before bulk entry prevents the most common and most expensive class of error, which is discovering inconsistencies after thousands of records have already been created.
New MDR/IVDR devices must have a complete device record before first placement on the market from 28 May 2026; legacy devices still marketed afterwards must be registered by 28 November 2026. For organisations with both, it is worth separating the two populations explicitly in the project plan, because they have different deadlines and often very different data-readiness levels.
Step 4: Link Notified Body Certificates
For devices requiring Notified Body involvement, the Notified Body links the relevant certificate to the registered device in EUDAMED. The manufacturer’s role here is to ensure the device record is complete and accurate so the link can be made without friction. Mismatched data between the device record and the certificate is a frequent source of delay at this stage, and because it involves a third party, the manufacturer cannot simply fix it unilaterally at the last moment. Confirm the certificate-upload timing and process with your Notified Body well ahead of the deadline rather than assuming it will happen automatically; Notified Bodies are managing this across their entire client base simultaneously, so early coordination is a competitive advantage in queue terms.
Step 5: Maintain and Keep Data Current
EUDAMED is not a one-time submission. Once registered, EUDAMED becomes the single source of truth, and the data must be kept accurate over the device lifecycle. Build EUDAMED maintenance into your quality system processes alongside post-market surveillance, vigilance and labelling work, so that any change that affects a registered attribute triggers a corresponding EUDAMED update through a controlled process rather than being remembered ad hoc. For manufacturers with large portfolios, manual entry will not scale: evaluate machine-to-machine (M2M) integration, which connects ERP/PLM systems directly to EUDAMED via the XML-based interface and is strongly recommended for bulk and ongoing data exchange. The decision to build M2M should be made early, because a validated machine interface is itself a project with its own lead time.
Machine-to-Machine Integration: When and Why
For a manufacturer with a handful of devices, manual data entry into EUDAMED is workable. For a manufacturer with hundreds or thousands of UDI-DIs, manual entry is both a deadline risk and an ongoing maintenance liability, and this is where machine-to-machine integration becomes a strategic rather than a technical decision.
M2M integration uses an XML-based interface to exchange data directly between the manufacturer’s ERP or PLM system and EUDAMED, allowing bulk creation and bulk maintenance of device records without manual transcription. The advantages are not only speed at the initial upload but accuracy and sustainability over the lifecycle: if the ERP/PLM system is the controlled master for device data, an automated interface keeps EUDAMED synchronised with it as products change, which directly addresses the data-consistency requirement that causes so many delays.
The trade-off is lead time and validation. An M2M connection requires a validated architecture, a test environment, appropriate security, and conformance to the prescribed exchange protocols. None of this is instantaneous, and a manufacturer that decides in early 2026 that it needs M2M for a large portfolio may find that the integration cannot be built, validated and populated before the deadline. The practical guidance is therefore to make the build-versus-manual decision as one of the first actions in the project, sized against the portfolio: the larger the device count and the more frequently it changes, the stronger the case for M2M and the more urgent the decision.
✦ COMPLETE CATALOG
Find the documentation you need — instantly.
Whether you need a complete kit or just one specific SOP, our catalog has it. 45 process packages and 3 complete bundles, all instantly downloadable and fully editable.
- ✓ Complete bundles or individual packages
- ✓ 45 process packages from €69 each
- ✓ ISO 13485 · MDSAP · Combined Kit
Common Registration Mistakes and How to Avoid Them
Several errors recur across organisations preparing for EUDAMED, and each is avoidable with foresight.
The first is starting with device data instead of actor registration. Because device records cannot be created without an SRN, any plan that front-loads device data preparation while leaving actor registration late has the dependency backwards. Actor registration, and for non-EU manufacturers the appointment of an EU authorised representative, must be the first action, not a parallel one.
The second is treating data consistency as a clean-up task at the end. Inconsistencies between EUDAMED, labelling, IFU, technical documentation and certificates are the dominant cause of delay, and they are far cheaper to prevent than to remediate. Establishing a single master source for each data element before bulk entry, as described above, is the structural defence.
The third is underestimating the legacy-device population. The 28 November 2026 deadline for devices already on the market is often the larger workload, because legacy products frequently lack the structured UDI data that newer products have, and creating it retrospectively across an old portfolio is slow. Treating the legacy population as a distinct, separately resourced workstream prevents it from being discovered late.
The fourth is assuming the Notified Body step will resolve itself. Certificate linking depends on a third party operating under its own capacity constraints across all its clients. Manufacturers who coordinate the certificate timeline with their Notified Body early avoid being in an undifferentiated queue close to the deadline.
The fifth is forgetting the supply-interruption obligation. Under Regulation (EU) 2024/1860 there is a separate obligation to notify authorities in advance of any interruption or discontinuation of supply for critical devices, and a device incorrectly left registered as “on the market” in EUDAMED can expose the manufacturer to additional liability. Data accuracy in EUDAMED therefore has direct legal weight beyond the registration formality itself, and the maintenance process must include keeping market-status fields truthful.
Practical Tips for a Tight Timeline
Three priorities separate organisations that will be ready from those that will not.
First, treat Actor registration as the immediate critical path — start it now, because the SRN gates everything and competent-authority validation takes time that is outside your control. For non-EU manufacturers, the appointment and formalisation of the EU authorised representative is the action that precedes even this.
Second, run a UDI/DI gap assessment across MDR, IVDR and legacy devices to know exactly how many device records you must create and by which of the two deadlines each falls under. You cannot resource the project realistically until you know the true size of the device population and how much of its data already exists in a usable form.
Third, decide early whether you need M2M integration. For a portfolio of any meaningful size, manual data entry into EUDAMED close to the deadline is a significant operational risk, and building a validated M2M connection is itself a project with lead time. The decision cannot be deferred until the manual approach has visibly failed.
A final point of consequence ties these together: EUDAMED is now a core input into portfolio and lifecycle planning, not a standalone IT task. It should sit alongside certification planning, clinical and performance evaluation, labelling and post-market surveillance, with its milestones embedded in the same programme. Organisations that integrate EUDAMED into their existing regulatory programme will absorb the transition; those that run it as an isolated, last-minute exercise carry the highest risk of losing market access at the cut-off.
✦ PREMIUM BUNDLE · EU MDR COMPLETE
The complete EU MDR documentation set — technical, clinical and risk.
The Technical Documentation Kit (Annex II), the Clinical Documentation Kit (Articles 32, 61, 83-86) and the Risk Management Kit (ISO 14971 + IEC 62366-1) in one premium bundle — the controlled sources every EUDAMED device record has to stay consistent with. 22 templates, cross-referenced, with a single Master Index.
- ✓ 22 templates · pre-market + post-market documentation
- ✓ Save €178 vs buying the three kits separately
FROM
€999
Get the MDR Complete Bundle →Frequently Asked Questions
When does EUDAMED become mandatory?
The four functional modules — Actor Registration, UDI/Device Registration, Notified Bodies and Certificates, and Market Surveillance — become mandatory on 28 May 2026, six months after Commission Decision (EU) 2025/2371 was published on 27 November 2025. From that date, obligations linked to these modules must be fulfilled in EUDAMED and parallel national systems no longer suffice.
What is an SRN and why does it matter?
The Single Registration Number is the unique identifier issued to an economic operator after Actor registration is validated by the competent authority. It is the gatekeeper of the whole system: without a valid SRN you cannot register devices or place them on the EU market. Obtaining the SRN should be the first action in any EUDAMED plan because validation is not instantaneous and the time it takes is outside the manufacturer’s control.
What are the deadlines for registering devices?
New MDR/IVDR devices must be registered before they are placed on the EU market from 28 May 2026. Devices already on the market before 28 May 2026 that continue to be marketed afterwards must be registered by 28 November 2026. Notified Bodies must complete the upload of relevant legacy certificate information by 28 May 2027.
Do non-EU manufacturers need to register in EUDAMED?
Yes. Non-EU manufacturers must register in the Actor module and must also have an EU authorised representative who confirms the registration. The authorised representative must be in place before registration can be completed, so this should be arranged well ahead of the deadline and is typically the first action a non-EU manufacturer should take.
Do distributors have to register in EUDAMED?
Distributors are not economic operators required to complete Actor registration in the same way as manufacturers, authorised representatives and importers. However, they have a practical obligation to verify that the operators they buy from are registered and that device data is consistent, and to maintain accessible documentation during the transition. Distributor verification procedures should be updated to include an EUDAMED check.
Is EUDAMED registration linked to the MDR/IVDR transition deadlines?
No. EUDAMED registration runs on its own schedule defined by Regulation (EU) 2024/1860 and the module-functionality notices, independent of the MDR/IVDR certificate transition periods. Meeting a certification deadline does not exempt an operator from the EUDAMED registration dates, which must be planned as a separate workstream.
Can I use machine-to-machine integration to register in bulk?
Yes. EUDAMED supports an XML-based machine-to-machine interface that lets manufacturers exchange data directly between their ERP/PLM systems and EUDAMED, which is strongly recommended for large portfolios. However, an M2M connection requires a validated architecture, a test environment and conformance to the prescribed protocols, so the decision to build it must be taken early because it has its own lead time.
Conclusions
EUDAMED registration has moved from a deferred project to an enforceable obligation with fixed, near-term dates. The practical takeaways are clear: appoint and formalise an EU authorised representative now if you are a non-EU manufacturer; submit Actor registration immediately because the SRN gates everything and validation takes time you do not control; map a single master source for every device data element before bulk entry so that EUDAMED, labelling, IFU, technical documentation and certificates are consistent by construction; separate and resource the legacy-device population explicitly against its 28 November 2026 deadline; coordinate certificate linking with your Notified Body early; decide the M2M question at the start of the project rather than after manual entry has failed; and treat EUDAMED as a maintained single source of truth whose accuracy now carries legal weight. Organisations that integrate these steps into their existing regulatory programme will absorb the transition; those that wait risk losing EU market access at the 28 May 2026 cut-off.
The EU MDR Compliance Documentation Package on MD Regulatory includes an EUDAMED readiness checklist, an actor registration data-gathering template, a UDI/DI portfolio gap-assessment worksheet, and a device-data consistency checklist aligned with the EUDAMED UDI/Device module — built to help manufacturers execute the steps in this guide against the 2026 deadlines.
Related articles