ISO 13485:2016 — The Complete Guide to Medical Device Quality Management
Table of Contents
ISO 13485 is the internationally recognized standard for quality management systems in the medical device industry. For any manufacturer designing, producing, installing, or servicing medical devices — anywhere in the world — ISO 13485 is not simply a best practice. It is the foundational framework that underpins market access in virtually every major regulatory jurisdiction, from the European Union under EU MDR to the United States under the newly enacted FDA Quality Management System Regulation (QMSR).
ISO 13485:2016 was last reviewed and confirmed in 2025, meaning this version remains current and fully applicable. This article provides the most comprehensive practical guide to ISO 13485 available — covering its scope, structure, clause-by-clause requirements, key differences from ISO 9001, relationship to EU MDR and FDA QMSR, and how to implement it effectively in a medical device organization.
AUDIT-READY KIT
Build your ISO 13485 QMS with confidence.
Built on 15+ years of audit experience — every SOP and template references the regulations auditors expect. Get to certification faster, with industry best practices baked in.
- ✓30 SOPs covering the full QMS scope
- ✓56 templates ready to customize
- ✓Aligned with EU MDR + FDA QMSR
What Is ISO 13485 and Why Does It Matter?
ISO 13485 is designed to be used by organizations involved in the design, production, installation and servicing of medical devices and related services. It can also be used by internal and external parties, such as certification bodies, to help them with their auditing processes.
At its core, ISO 13485 defines what a quality management system must look like for a medical device organization — the processes, documentation, responsibilities, and controls that must be in place to consistently produce safe and effective devices. Unlike general quality standards, ISO 13485 is specifically designed around the regulatory requirements of the medical device industry, with an emphasis on risk management, process validation, regulatory compliance, and product traceability throughout the entire lifecycle.
Why it matters in practice?
ISO 13485 certification is either directly required or strongly expected as a prerequisite for market access in the EU (under EU MDR 2017/745), Canada (under MDSAP), Japan (JPAL), Brazil (ANVISA), and Australia (TGA). In the United States, the FDA’s Quality Management System Regulation (QMSR), which became effective on February 2, 2026, incorporates ISO 13485:2016 by reference into 21 CFR Part 820, making it the legal basis for FDA quality system inspections of medical device manufacturers.
In practical terms this means that a single ISO 13485-compliant QMS can now satisfy quality system requirements across multiple major markets simultaneously — dramatically reducing the regulatory burden for manufacturers operating globally.
ISO 13485 QMS Documentation Bundle
Building a fully compliant ISO 13485 QMS requires not only a thorough understanding of the standard’s requirements, but also a complete set of audit-ready documentation covering every core process — from risk management and design controls to CAPA, supplier management, and post-market surveillance. The ISO 13485 QMS Documentation Bundle provides over 70 fully editable Word and Excel templates covering 22 QMS processes, written by regulatory professionals with direct audit experience and structured to reflect what Notified Bodies and FDA inspectors expect to see. For manufacturers building their QMS from scratch, preparing for a certification audit, or standardizing an existing system, this bundle eliminates weeks of documentation work and significantly reduces the risk of nonconformities at audit.
ISO 13485 vs ISO 9001 — Key Differences
ISO 13485 and ISO 9001 are both quality management system standards, but they serve fundamentally different purposes and should not be confused.
ISO 13485 addresses unique medical device needs that ISO 9001 simply cannot handle: regulatory compliance focus rather than customer satisfaction emphasis, risk management integration throughout all processes, validation requirements for processes affecting product safety, and detailed documentation supporting regulatory submissions.
The most significant structural differences are:
Regulatory focus vs customer focus. ISO 9001 is built around customer satisfaction and continual improvement. ISO 13485 is built around regulatory compliance and consistent product safety — the primary obligation is to regulators and patients, not commercial customers.
Risk-based approach. Both standards incorporate risk-based thinking, but ISO 13485 goes substantially further — requiring specific risk management processes for product design, manufacturing, and post-market activities, aligned with ISO 14971.
Continual improvement. ISO 9001 requires continual improvement of the QMS itself. ISO 13485 requires only the maintenance of the QMS effectiveness — a meaningful distinction that reflects the conservative, stability-oriented nature of medical device regulation.
Sterile product and implantable device requirements. ISO 13485 contains specific clauses covering sterile product manufacturing, implantable devices, and associated documentation requirements that have no equivalent in ISO 9001.
Documentation and records. ISO 13485 imposes significantly more rigorous documentation requirements — manufacturers must maintain documented procedures for every process that affects product safety and quality, with records retained for periods defined by regulatory requirements.
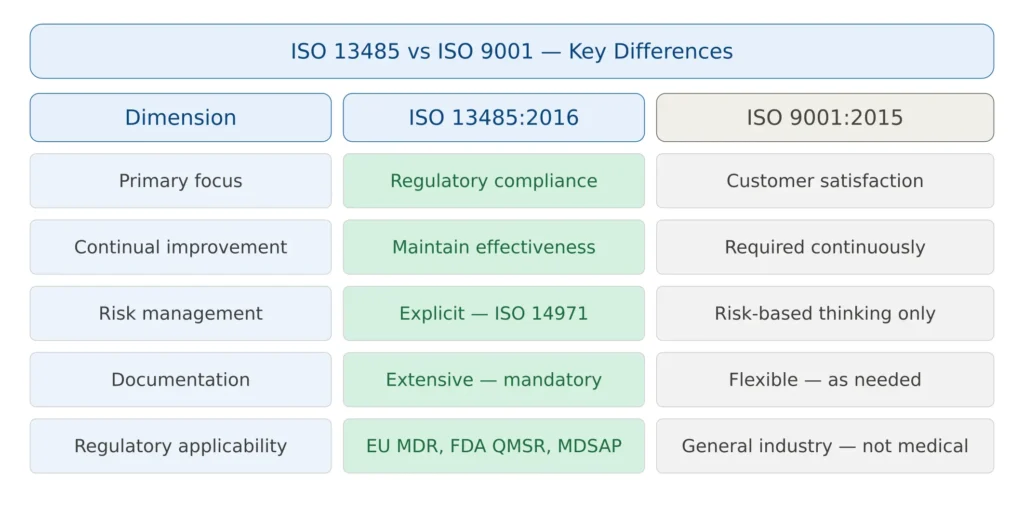
Figure 1 — ISO 13485 vs ISO 9001: key structural differences
ISO 13485 Structure — The Seven Clauses
ISO 13485:2016 is organized into seven main clauses, following a structure similar to other ISO management system standards. Clauses 1-3 cover scope, normative references, and terms — the operational requirements begin at Clause 4.
Clause 4 — Quality Management System (General Requirements)
This clause establishes the foundational requirements for the QMS as a whole. Manufacturers must determine the processes needed for the QMS, their sequence and interaction, the criteria and methods for ensuring their effective operation, and the resources needed to support them. It also introduces the concept of risk-based approach to quality management — requiring that processes are controlled in proportion to the risk they pose to product safety and quality.
A key element of Clause 4 is the Quality Manual — a documented description of the QMS scope, the documented procedures or references to them, and the interaction between the QMS processes. The Quality Manual is a mandatory output of Clause 4 under ISO 13485 (unlike ISO 9001:2015, which removed this requirement).
Clause 5 — Management Responsibility
Top management — not just the quality department — must demonstrate active commitment to the QMS through:
Quality policy: A documented statement of the organization’s commitment to quality, appropriate to the organization’s purpose, providing a framework for quality objectives, and reviewed for continuing suitability.
Quality objectives: Measurable, specific objectives established at relevant functions and levels of the organization, consistent with the quality policy.
Management review: Periodic structured reviews of the QMS by top management, assessing its continuing suitability, adequacy, and effectiveness. The management review must cover defined inputs (including audit results, customer feedback, process performance, CAPA status, regulatory changes, and recommendations for improvement) and produce defined outputs (decisions on resource needs, process improvements, and product improvements).
Person Responsible for Regulatory Compliance (PRRC): Under EU MDR 2017/745, manufacturers must designate a PRRC — the individual responsible for ensuring regulatory compliance. ISO 13485 Clause 5 aligns with this requirement through its management representative provisions.
✦ PREMIUM BUNDLE
The ultimate global QMS documentation bundle.
Combine ISO 13485 + all 5 MDSAP markets in one premium package. Deduplicated structure means you customize each document once — not twice. Save €199 vs buying the kits separately.
- ✓41 SOPs covering both ISO 13485 and MDSAP
- ✓70+ templates with deduplicated structure
- ✓Save €199 vs buying separately
Clause 6 — Resource Management
Manufacturers must determine and provide the resources needed to implement and maintain the QMS and continually improve its effectiveness. This covers:
Human resources: Personnel performing work affecting product quality must be competent on the basis of education, training, skills, and experience. Competence must be documented and records maintained.
Infrastructure: Buildings, workspace, equipment, supporting services (IT, communication) — all must be appropriate to achieving conformity to product requirements.
Work environment: The physical conditions of the work environment must be managed to the extent necessary to achieve product conformity, including cleanliness requirements, contamination control, and environmental monitoring where applicable.
Clause 7 — Product Realization
This is the most operationally detailed clause of ISO 13485 — covering everything from design to delivery. Its subclauses address:
7.1 — Planning of product realization: For each product or product family, manufacturers must plan the quality activities, verification and validation activities, responsibilities, and documentation needed to deliver a conforming product.
7.2 — Customer-related processes: Determination of product requirements, including applicable regulatory requirements, review of requirements before commitment to supply, and communication with customers on product information, feedback, and complaints.
7.3 — Design and development: One of the most critical and frequently audited subclauses. It covers design planning, design inputs, design outputs, design review, design verification, design validation, design transfer, design changes, and the Design History File (DHF). For software, design and development must also address the requirements of IEC 62304.
7.4 — Purchasing: Suppliers and externally provided processes, products and services must be controlled. The extent of control must be proportionate to the risk to product safety and quality — critical suppliers require more rigorous qualification and monitoring than non-critical ones.
7.5 — Production and service provision: Manufacturing process control, cleanliness requirements, installation activities, servicing activities, and the specific requirements for sterile medical devices and implantable devices.
7.6 — Control of monitoring and measuring equipment: All measurement equipment used to verify product conformity must be calibrated at specified intervals, adjusted as necessary, identified, safeguarded, and records maintained.
Clause 8 — Measurement, Analysis and Improvement
This clause covers the monitoring and measurement activities that provide evidence that the QMS is working as intended:
8.1 — General: Planning and implementation of monitoring, measurement, analysis, and improvement processes needed to demonstrate conformity of the product, ensure conformity of the QMS, and maintain its effectiveness.
8.2 — Monitoring and measurement: Including customer feedback and complaint handling, internal audit, monitoring and measurement of processes, and monitoring and measurement of product.
8.3 — Control of nonconforming product: A documented procedure is required for identifying, documenting, segregating, evaluating, and disposing of nonconforming product. This applies to incoming nonconforming materials, in-process nonconformances, and final product that fails to meet requirements.
8.4 — Analysis of data: The organization must determine, collect, and analyze appropriate data to demonstrate the suitability and effectiveness of the QMS and to evaluate improvements — including data from feedback, audits, process monitoring, and supplier performance.
8.5 — Improvement: Including CAPA (Corrective and Preventive Action) — arguably the most scrutinized element in regulatory audits. The CAPA process must identify the root cause of nonconformities, implement corrections and corrective actions, verify their effectiveness, and record results.
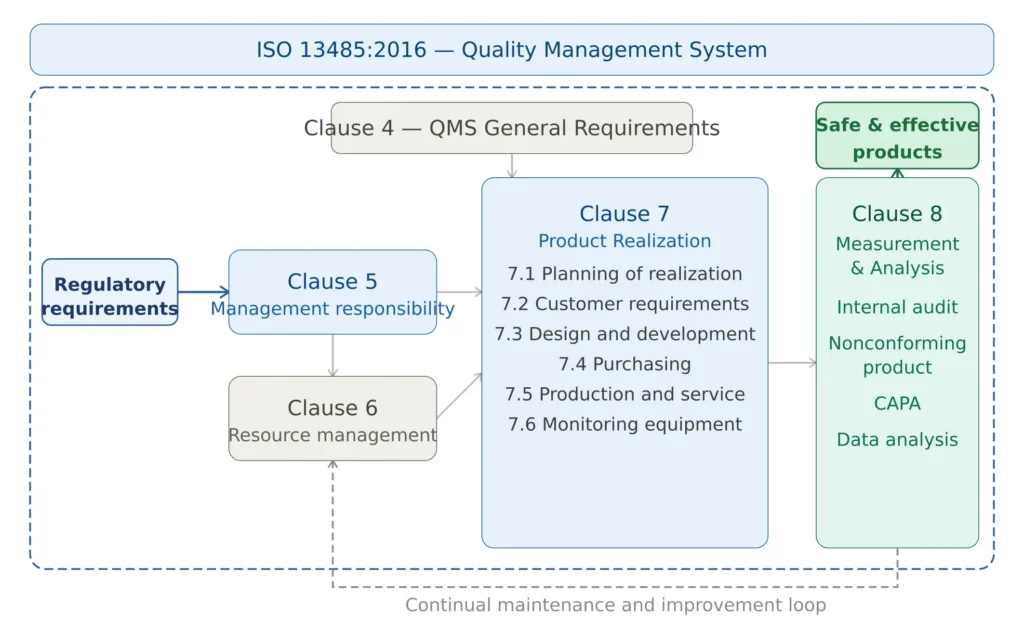
Figure 2 — ISO 13485 structure: seven clauses and key process interactions
ISO 13485 and the FDA QMSR — A Historic Alignment
One of the most significant developments in medical device quality management in recent years is the alignment of the FDA’s quality system requirements with ISO 13485. The Quality Management System Regulation (QMSR) that became effective on February 2, 2026, amends the device current good manufacturing practice requirements of 21 CFR Part 820, incorporating by reference ISO 13485:2016 Medical devices — Quality management systems — Requirements for regulatory purposes. This action harmonizes the FDA’s CGMP regulatory framework with that used by other regulatory authorities.
The practical implications of this are profound. For years, manufacturers selling in both US and international markets have maintained two separate quality systems — one for FDA compliance and another for ISO certification. The new QMSR eliminates much of that duplication by aligning both systems.
However, the QMSR is not identical to ISO 13485. FDA inspections under the QMSR will not follow the MDSAP audit plan or procedures. The FDA will not require certificates of conformance to ISO 13485 and will not issue certificates of conformance to ISO 13485. A certificate of conformance to ISO 13485 will not exempt a manufacturer from an FDA inspection.
Key differences to be aware of:
Additional FDA-specific requirements: The QMSR adds specific requirements beyond ISO 13485 for labelling controls, packaging, UDI compliance, and certain documentation provisions that reflect FDA-specific expectations.
Inspection scope: FDA inspectors will now be able to review internal audits, supplier evaluations, and management review records that were previously exempt from inspection under the old QSR.
New inspection methodology: On February 2, 2026, the FDA stopped using the Quality System Inspection Technique (QSIT) for device inspections and began utilizing the inspection process described in the updated Inspection of Medical Device Manufacturers Compliance Program 7382.850.
For manufacturers already ISO 13485 certified, QMSR compliance is largely achieved — but a gap analysis focusing on the FDA-specific additions is still recommended.
COMPLETE CATALOG
Find the documentation you need — instantly.
Whether you need a complete kit or just one specific SOP, our catalog has it. 45 process packages and 3 complete bundles, all instantly downloadable and fully editable.
- ✓Complete bundles or individual packages
- ✓45 process packages from €69 each
- ✓ISO 13485 · MDSAP · Combined Kit
ISO 13485 and EU MDR — How They Work Together
ISO 13485 and EU MDR 2017/745 are complementary but distinct frameworks. ISO 13485 defines how a quality management system must be structured and operated. EU MDR defines what must be demonstrated about specific medical devices and their conformity with safety and performance requirements.
In practice, ISO 13485 certification is not legally required by EU MDR — but it is the most efficient and universally recognized way to satisfy the QMS requirements of MDR Article 10(9) and Annex IX. Notified Bodies use ISO 13485 as their primary reference when auditing manufacturer QMS under MDR conformity assessment procedures.
The key points of interaction between ISO 13485 and EU MDR are:
Design controls (Clause 7.3) directly feed into EU MDR Annex II technical documentation requirements — the Design History File is the bridge between the QMS and the technical file.
Post-market surveillance processes required by MDR Articles 83-86 must be embedded in the QMS under ISO 13485 Clause 8 — as data sources for management review, triggers for CAPA, and inputs to the risk management file.
Supplier management (Clause 7.4) maps to EU MDR’s requirements for economic operator obligations and supply chain control, as well as to the SOUP management requirements of IEC 62304 for software components.
Management review (Clause 5.6) must incorporate EU MDR-specific inputs including EUDAMED data, vigilance reports, and regulatory change monitoring.
Key ISO 13485 Documentation Requirements
One of the areas where ISO 13485 is most demanding — and most frequently found deficient in audits — is documentation. The standard requires a specific set of documented procedures and records as mandatory minimum outputs.
Mandatory documented procedures include: control of documents, control of records, internal audit, control of nonconforming product, corrective action, and preventive action. These six documented procedures are explicitly required regardless of device class or organization size.
Beyond the mandatory six, ISO 13485 requires documented procedures for any process where their absence could adversely affect quality — which in practice means most core QMS processes require documentation.
Records that must be maintained include: management review outputs, education and training records, evidence of product realization planning, results of design and development activities, results of purchasing evaluations, calibration records, complaint records, internal audit reports, nonconforming product records, corrective and preventive action records.
The quality management system documentation hierarchy typically consists of four levels: the Quality Manual (Level 1), Standard Operating Procedures (Level 2), Work Instructions (Level 3), and Forms and Records (Level 4). All documents must be controlled through a formal document control procedure per Clause 4.2.4.
Most Common ISO 13485 Audit Findings
Based on industry experience and published Notified Body data, these are the most frequently cited nonconformities in ISO 13485 audits — both by Notified Bodies under EU MDR and by FDA inspectors under QMSR.
The most frequently cited QMS nonconformities include incomplete design control procedures — missing or inadequate design input/output requirements, design verification and validation plans, or design transfer procedures. Also common is inadequate risk management integration — risk management treated as a separate exercise rather than integrated throughout the product lifecycle from design inputs through post-market surveillance. Software validation gaps are also frequent — software used in the quality management system not validated to the level required. Supplier management weaknesses are another common finding — inadequate supplier qualification, monitoring, or control, particularly for critical suppliers and contract manufacturers.
Additional frequent findings include CAPA systems that identify problems but do not adequately verify effectiveness of corrective actions, management reviews that exist on paper but do not demonstrate genuine top management engagement with QMS data, and complaint handling processes that are reactive rather than proactive in identifying trends.

Figure 3 — Most common ISO 13485 audit findings
ISO 13485 Implementation — A Practical Roadmap
Implementing ISO 13485 from scratch — or upgrading an existing QMS to full compliance — is a substantial project. The following roadmap reflects industry best practice for organizations of small to medium size.
Phase 1 — Gap Analysis and Planning (Months 1-2)
The starting point is always a structured gap analysis comparing your current practices against each clause of ISO 13485. The gap analysis should produce a prioritized list of missing or inadequate elements, an implementation plan with resource allocation and timelines, and a decision on certification scope — which sites, which product lines, which processes are included.
If you are transitioning from the old FDA QSR to QMSR, the gap analysis should focus specifically on the ISO 13485 requirements that were not present in the QSR — particularly design controls for Class I devices (now required under QMSR), and the expanded documentation requirements.
Phase 2 — Documentation Development (Months 2-5)
Develop or update all required documented procedures, work instructions, forms, and templates. Start with the six mandatory procedures (document control, record control, internal audit, nonconforming product, CAPA, preventive action), then build outward to cover all core processes.
A common mistake is writing procedures that are too long and too detailed — procedures become unwieldy and staff don’t follow them in practice. The best ISO 13485 procedures are concise, process-oriented, and clearly written for the people who will actually use them.
Phase 3 — Implementation and Training (Months 4-7)
Deploy the new or updated procedures across the organization. Training must be documented and verified — not just a briefing session, but evidence that staff understand and can apply the requirements in their daily work. Internal training records are one of the first things Notified Bodies and FDA inspectors review.
Phase 4 — Internal Audit (Months 6-8)
Before submitting to a certification body, conduct at least one full internal audit cycle covering all clauses of ISO 13485 in scope. The internal audit must be conducted by trained auditors who are independent of the processes being audited. Findings from the internal audit must be addressed through the CAPA system.
Phase 5 — Management Review and Readiness (Month 8-9)
Conduct a formal management review covering all required inputs. This is often the last step before the certification audit — it demonstrates to the Notified Body that the QMS is operating, generating data, and being actively overseen by top management.
Phase 6 — Certification Audit
The certification body conducts a two-stage audit: Stage 1 (documentation review, typically remote) and Stage 2 (on-site audit of QMS implementation). Any major nonconformities must be resolved before the certificate is issued. Minor nonconformities must have a corrective action plan. After certification, annual surveillance audits and a three-year recertification cycle maintain the certificate.

Complete Bundle
ISO 13485 QMS Documentation Bundle — 70+ Editable Templates
Everything you need to build a fully compliant ISO 13485 QMS — 70+ documents covering 22 processes. Instantly downloadable in Word and Excel format.
ISO 13485 and MDSAP — The Global Audit Program
The Medical Device Single Audit Program (MDSAP) deserves specific mention because it represents one of the most powerful efficiency tools available to manufacturers operating in multiple markets. MDSAP is a voluntary program that allows a single audit conducted by an authorized MDSAP auditing organization to satisfy the regulatory audit requirements of Australia (TGA), Brazil (ANVISA), Canada (Health Canada), Japan (MHLW/PMDA), and the United States (FDA).
MDSAP is a voluntary third-party audit program and will still be available to manufacturers who choose to participate under the QMSR framework. The MDSAP audit covers ISO 13485 requirements plus the specific regulatory requirements of each participating country — making it particularly valuable for manufacturers with multi-market strategies.
For manufacturers already ISO 13485 certified, the incremental effort to achieve MDSAP certification is manageable — primarily the addition of country-specific regulatory requirements for each target market. The return on investment is significant: one audit instead of up to five separate national audits.
The Future of ISO 13485 — What’s Coming
ISO 13485 is currently under review. It is considered that changes related to the new ISO Harmonized Structure, including a focus on climate change, that have been implemented for other management system standards may be considered. Additionally, the integration of new technologies like AI into medical devices may lead to future amendments or new editions of ISO 13485. It is currently understood that the updated version will be published in September 2026.
Manufacturers should begin monitoring the revision process and assessing potential impacts on their QMS — particularly around AI governance, climate-related sustainability requirements, and any structural changes from the ISO Harmonized Structure adoption. The current 2016 version remains fully valid and enforceable, and the FDA has explicitly noted that future ISO 13485 revisions will not automatically apply to QMSR without new rulemaking.
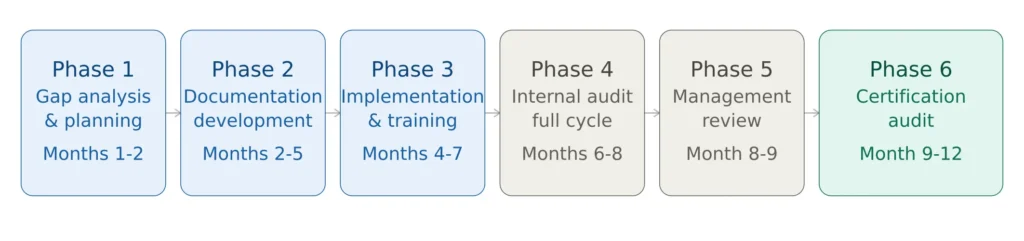
Figure 4 — ISO 13485 implementation roadmap
Frequently Asked Questions on ISO 13485
Is ISO 13485 certification mandatory for EU MDR compliance? ISO 13485 certification is not explicitly mandated by EU MDR 2017/745, but it is the universally accepted means of demonstrating compliance with the QMS requirements of MDR Article 10(9). In practice, every Notified Body uses ISO 13485 as the reference framework for QMS audits under MDR conformity assessment. For Class IIa, IIb, and III devices, Notified Body QMS assessment is mandatory — and without ISO 13485 compliance there is no realistic path to CE marking.
Does ISO 13485 certification replace FDA inspection? No. A certificate of conformance to ISO 13485 will not exempt a manufacturer from an FDA inspection. The QMSR incorporates ISO 13485 by reference as the legal standard, but FDA compliance is assessed through FDA inspections — not through certification body audits.
How long does ISO 13485 certification take? For most small to medium medical device companies, the full implementation and certification process takes 9 to 18 months depending on the starting maturity of the QMS, the scope of certification, and the availability of a certification body. Organizations already operating under ISO 9001 or the old FDA QSR typically require less time.
What is the difference between ISO 13485 and MDSAP? ISO 13485 is the standard. MDSAP is an audit program that uses ISO 13485 as its reference, adding the specific regulatory requirements of five participating countries (Australia, Brazil, Canada, Japan, USA). MDSAP certification satisfies both ISO 13485 certification and the regulatory audit requirements of all five countries simultaneously.
Will ISO 13485 be updated? It is currently understood that an updated version will be published in September 2026. The current 2016 version remains fully valid. Manufacturers should monitor the revision process but do not need to take immediate action — transition periods will be provided when a new version is published.
Conclusions
ISO 13485 is the cornerstone of medical device quality management worldwide. With its incorporation into the FDA QMSR in February 2026, it has now become the single most important quality management standard for any manufacturer selling medical devices globally — satisfying regulatory expectations across the EU, US, Canada, Japan, Brazil, and Australia within a single unified framework.
Building and maintaining an ISO 13485-compliant QMS is not a one-time project — it is a continuous operational discipline that must be embedded into the daily work of every function involved in the design, production, and post-market lifecycle of medical devices. Organizations that treat it as a living system rather than a documentation exercise consistently achieve better audit outcomes, fewer regulatory findings, and faster market access.
For manufacturers still in the process of implementing ISO 13485, the most valuable first investment is a thorough gap analysis — understanding exactly where your current practices fall short of the standard’s requirements, and building a realistic implementation plan from there.
This article is part of the MD Regulatory series on medical device compliance. Related articles cover EU MDR technical documentation requirements, ISO 13485 internal audit checklist, CAPA procedure under ISO 13485, and FDA QMSR 21 CFR Part 820.