ISO 10993-1:2025 – A Breakthrough Update Reshaping Medical Device Biocompatibility
Table of Contents
Introduction
The sixth edition of ISO 10993-1 introduces updated requirements that affect both the biological evaluation process and the assessment of medical devices during development and throughout their lifecycle, including those already placed on the market.
This sixth edition represents a significant shift in certain aspects of the biological evaluation process, while retaining most established practices. However, these changes will impact how manufacturers conduct biological evaluations and how assessors, including FDA and EU Notified Bodies, review them.
In this article we will discuss the details related to this new edition of ISO 10993-1, focusing on the key changes and related impact to the medical device industry.
These changes in the ISO 10993-1 will have a concrete impact on medical devices, especially in regulatory submissions such as 510k process for US market or in the framework of QMS certification such as MDSAP.
Summary of the Key Changes
Compared to the previous edition, several significant updates have been introduced within the new version of ISO 10993-1. A major structural revision has been implemented, with the document fully reorganised and its title aligned with the risk management principles defined in ISO 14971. In addition, Annex A has been streamlined to focus exclusively on materials characterization, while its previous content has been redistributed into the main body of the standard. A new Annex B has also been introduced to provide the rationale behind the revised classification of biological effects presented in Tables 1 to 4.
Further changes of ISO 10993-1 include the refinement of terminology and concepts used throughout the standard. The concept of “biological end points” has been revised and is now referred to as “biological effects” reflecting an updated approach to hazard identification. Similarly, the term “externally communicating” has been replaced with terminology that more precisely describes tissue contact at the level of device components. The expression “effects after implantation” has also been updated to “local effects after tissue contact”, recognising that such assessments may be required for devices that are not implanted.
Additional guidance has been incorporated to strengthen key aspects of the biological evaluation process, including more detailed direction on the characterization of medical devices, the identification of biological hazards, and the determination of exposure duration.
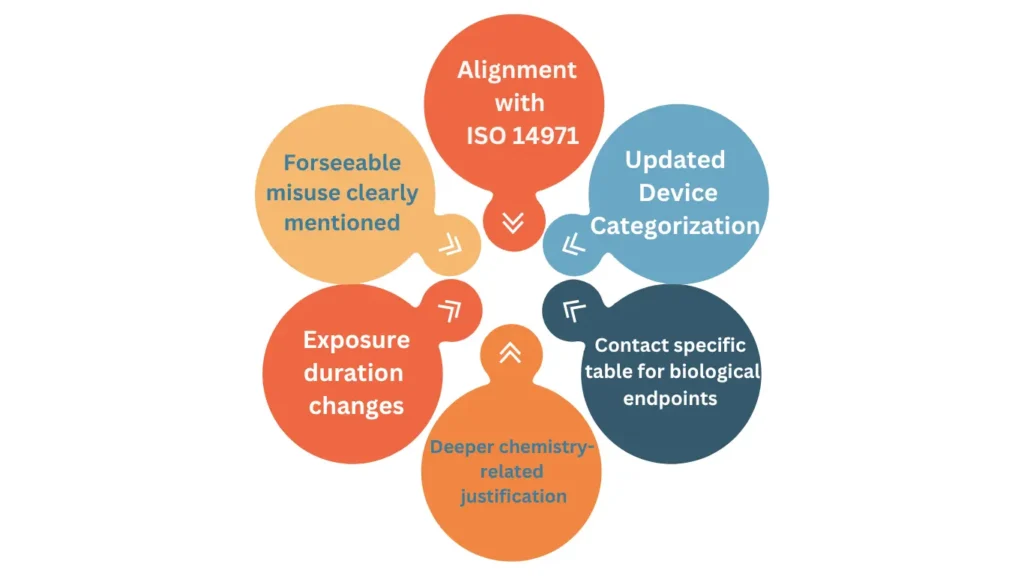
Impact on Device Categorisation
Several important updates have been introduced regarding terminology and classification criteria. Notably, the term ‘intact mucosal membrane’ has replaced the previous ‘mucosal membrane’, providing greater specificity in defining tissue conditions. In parallel, the former category ‘breached or compromised surface’ has been expanded and clarified as ‘breached or compromised surfaces or internal tissues other than blood’, ensuring a more comprehensive description of tissue interactions.
The classification framework has also been simplified through consolidation of categories. The concept of ‘externally communicating medical devices’ has been removed, with such devices now incorporated into either the category of devices contacting breached or compromised surfaces or internal tissues (excluding circulating blood), or into the blood-contacting category, as appropriate. Similarly, previously distinct classifications such as ‘tissue/bone/dentin’ and ‘tissue/bone’ are now encompassed within the broader category of breached or compromised surfaces or internal tissues other than blood. In addition, terms including ‘blood path, indirect’, ‘circulating blood’, and ‘blood’ have been harmonised under the single designation ‘circulating blood’.
A significant methodological update has also been introduced in relation to the determination of contact duration. The revised approach adopts the concept of ‘contact days’, whereby the number of days on which contact occurs is considered more relevant than the duration of contact within a single day. This change is particularly impactful for devices with transient yet repeated or cumulative exposure, as well as those involving intermittent contact, and is expected to influence categorisation decisions. Further illustrative examples are provided in Annex A of the ISO 10993-1 version 2025.
Redefinition of Biological Endpoints
The terminology and structure relating to biological evaluation have been significantly revised within this version of ISO 10993-1. The concept previously referred to as ‘biological endpoints’ has been replaced with ‘biological effects’, reflecting a broader and more risk-based perspective. In addition, the former endpoints table contained in Annex A has been removed and replaced by four separate tables integrated into the main body of the standard. These tables are further supported by supplementary considerations that extend beyond their content. Importantly, the standard explicitly clarifies that these tables are not intended to serve as prescriptive testing checklists, but rather as a framework for identifying and evaluating relevant biological effects within a risk management approach.
Classification of biological effects
Substantial updates have also been made to the classification and interpretation of biological effects. In the version 2025 of ISO 10993-1, the various categories of systemic toxicity (acute, subacute, subchronic, and chronic) have been consolidated into a single ‘systemic toxicity’ category. The term ‘irritation or intracutaneous reactivity’ has been simplified to ‘irritation’, while ‘implantation effects’ are now addressed under ‘local effects after tissue contact’. Furthermore, material-mediated pyrogenicity has been removed from the core list of biological effects. This change reflects a shift towards a more targeted approach, whereby testing for pyrogenicity is expected primarily in cases involving insufficiently characterised or novel materials.
Assessment of Systemic Toxicity
Additional expectations have been introduced regarding the evaluation of specific effects based on device characteristics. For all device categories beyond intact skin contact, manufacturers are expected to assess systemic toxicity in light of existing data. The consideration of genotoxicity has also been expanded, including for devices with breached or compromised tissue contact of certain durations, as well as for devices involving circulating blood exposure, where such assessments may now be required depending on contact conditions.
Finally, the ISO 10993-1 standard introduces an explicit requirement to evaluate additional criteria outlined in Clause 6.5.10, which are not fully captured within Tables 1 to 4. These sub-clauses define applicability based on factors such as contact type, duration, and material composition, reinforcing the need for a comprehensive and case-by-case biological evaluation.
ISO 10993-1 and Risk Management
The revision of the title of the ISO 10993-1 version 2025, together with the broader content updates, reflects a clear intention to align the biological evaluation process more closely with the risk management framework defined in ISO 14971. This alignment is evident through the increased use of risk-based terminology, the restructuring and renaming of clauses, and the introduction of process diagrams illustrating how biological evaluation integrates within the overall risk management lifecycle.
The updated ISO 10993-1 standard also introduces several key risk management concepts that were previously less explicit. These include the consideration of reasonably foreseeable misuse, the definition of risk acceptability criteria during the planning phase, and the evaluation of biological risk across the entire lifecycle of the device. In addition, greater emphasis is placed on the formal estimation and justification of biological risk as part of the overall assessment.
These changes represent a significant shift in approach, particularly for manufacturers who have historically treated biological evaluation as a checklist-driven activity or who have not fully integrated it within their risk management processes. The revised framework requires a more structured, risk-based, and lifecycle-oriented methodology, consistent with modern regulatory expectations.
What is the Impact of the New ISO 10993-1
It is unlikely that the introduction of these changes of 10993-1 will materially improve the overall safety profile of medical devices. As a result, industry adoption may be cautious rather than enthusiastic, particularly where the perceived regulatory burden increases without a clear corresponding safety benefit.
The real impact of the new edition is better understood by considering how it affects different dimensions of a manufacturer’s activities.
From a systems perspective, organizations will need to reassess their existing biological evaluation frameworks against the updated expectations of ISO 10993-1. This is not a minor update: it involves revisiting terminology, revising device characterization approaches, reinterpreting which biological effects are relevant, and—critically—ensuring proper alignment with the risk management principles defined in ISO 14971. In practice, this often translates into a structured gap analysis followed by procedural updates.
✦ PREMIUM BUNDLE
The ultimate global QMS documentation bundle.
Combine ISO 13485 + all 5 MDSAP markets in one premium package. Deduplicated structure means you customize each document once — not twice. Save €199 vs buying the kits separately.
- ✓41 SOPs covering both ISO 13485 and MDSAP
- ✓70+ templates with deduplicated structure
- ✓Save €199 vs buying separately
Impacts on the devices under Development
For devices that are already commercially available, the standard does not impose an automatic requirement for additional testing, provided safety and performance have already been adequately demonstrated. However, there is a clear expectation that existing biological evaluation reports will be critically reviewed. This review of ISO 10993-1 should verify whether the original assessment remains appropriate in light of updated characterization requirements, revised definitions of biological effects, and any newly introduced considerations. In some cases, additional endpoints may need to be addressed, although a well-documented history of safe clinical use may still be sufficient to justify the absence of further testing. The depth of this retrospective review will depend on the specific regulatory jurisdiction.
For products still in development, the implications vary depending on their stage of maturity. Devices approaching submission may require only limited adjustments, similar to those applied to marketed products. In contrast, early-stage developments should fully incorporate the revised approach, particularly in defining the future biological evaluation strategy. Timing of regulatory submissions and target markets will play a key role in determining whether immediate alignment with the new framework is necessary.
Impact on different Countries / Regulations
Geographical considerations are likely to be a decisive factor in how impactful these changes truly are. Different regulatory authorities are expected to adopt and interpret the new edition in varying ways, creating a fragmented global landscape. For example, Australia has actively contributed to several of the revisions and is expected to adopt the standard in a comprehensive manner. In Europe, alignment is anticipated through the harmonisation process following publication of the EN ISO version, although some divergence in opinion was observed during the approval phase. Canada is expected to follow a similar path to the EU. Japan, on the other hand, has indicated the possibility of developing a distinct national approach to biological evaluation and it may be possible it will not recognise iso 10993-1 version 2025. In the United States, the reception has been more critical, with stakeholders expressing reservations; any future recognition of ISO 10993-1 by the FDA is therefore likely to be accompanied by specific limitations, clarifications, or supplementary guidance.
Overall, the significance of this new edition lies less in immediate safety improvements and more in the shift toward a structured, risk-based, and globally variable approach to biological evaluation.
Is it necessary to perform Additional Testing?
The sixth edition of ISO 10993-1 clearly indicates that it does not impose an automatic obligation to perform additional testing. However, it does introduce the expectation that current biological evaluation practices and previously completed assessments are carefully reviewed in light of the revised framework.
Such a review may reveal gaps or misalignments that need to be addressed. Depending on the outcome, manufacturers may need to supplement their existing evidence base, either by sourcing additional information—such as published literature or data from relevant databases—or by generating new evidence through activities like enhanced chemical characterisation or targeted biological testing.
Reasonable Forseeable Misuse to be Taken in Consideration
The ISO 10993-1 standard states that device classification must take into account the possible consequences of reasonably foreseeable misuse, with particular attention given to patterns of misuse that emerge consistently from data analysis, such as findings from post-market surveillance activities. In essence, this requirement calls for a thoughtful evaluation of misuse scenarios—specifically, an assessment of how such misuse could influence the device’s classification, as well as its associated biological effects and safety profile.
However, this obligation should not be interpreted as requiring the device to be automatically classified according to misuse conditions, nor does it impose a requirement that risks arising from such misuse must be rendered acceptable through design modifications or reclassification. Rather, these aspects are intended to inform the manufacturer’s internal risk management and decision-making processes. They represent considerations to be weighed, not prescriptive outcomes that must be enforced.
Importantly, this nuanced interpretation is not explicitly articulated within the wording of the standard itself. This lack of clarity has been identified by some stakeholders as a shortcoming of the 6th edition, which they view as insufficiently developed in this area and open to divergent interpretations.
What about Transition Period)
ISO will establish a formal withdrawal date for the previous edition—typically allowing a transition period of around three years before ISO 10993-1 version 2018 is officially superseded. However, ISO itself does not impose or control regulatory timelines for compliance with its standards. Instead, the responsibility for defining transition expectations lies with regulatory authorities, which may specify a period during which they continue to accept biological evaluations conducted under the 2018 version.
In practice, experience shows that conformity expectations can differ significantly across EU Notified Bodies. Some may begin requiring alignment with the 6th edition relatively early, while others may adopt a more gradual approach. This variability reflects a degree of subjectivity in interpretation and enforcement. While manufacturers are justified in challenging overly aggressive expectations, it is generally more effective to do so from a position of preparedness—namely, by having a clear and documented transition strategy toward compliance with the new edition already in place.
General Flowchart for Biological Assessment According to ISO 10993-1 version 2025
The flowchart depicted in the referenced image provides a structured approach for determining the exposure duration of a medical device in accordance with ISO 10993‑1 version 2025, guiding manufacturers in evaluating how the device interacts with the body over time and the potential implications for biocompatibility testing and risk assessment.
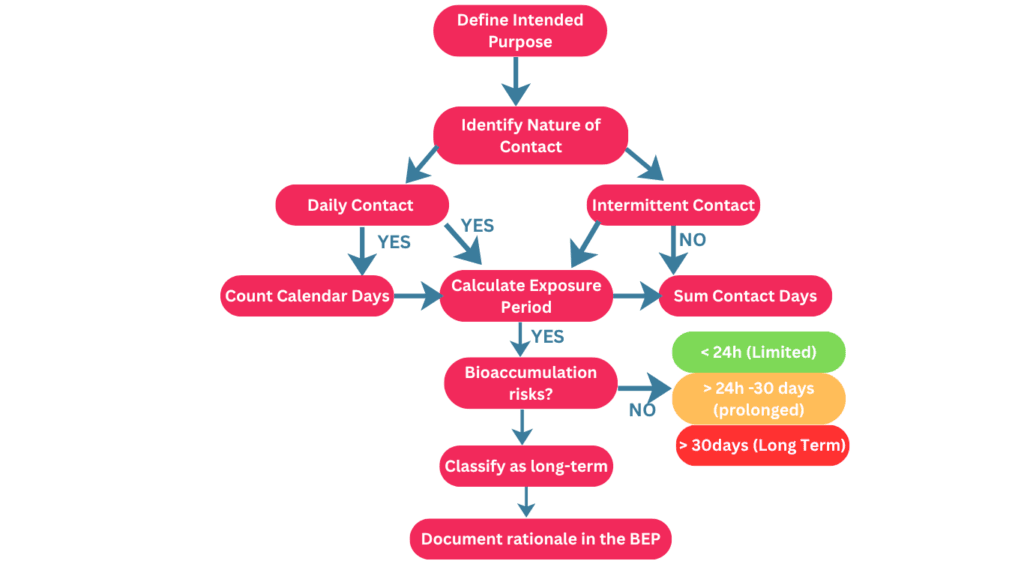
It begins with defining the intended use of the device, including the patient population, duration of each use, and frequency, which collectively establish the baseline for exposure analysis. The next step considers the nature of the device’s contact—whether it is surface-contacting, externally communicating, or implanted—which influences the severity of potential biological effects. The flowchart then directs evaluation of the frequency of contact, distinguishing between daily and intermittent use, with calculations of total exposure days reflecting either consecutive calendar days or the accumulation of separate use events. An additional consideration is the potential for bioaccumulation of device materials, which may override simple exposure duration and necessitate classification as long-term exposure due to persistent chemicals or substances. Based on these assessments, the device is categorized as limited, prolonged, or long-term exposure, ensuring that the biocompatibility testing strategy is appropriate for the level and nature of contact. The flowchart emphasizes that this process is both analytical and documentation-driven, requiring manufacturers to justify exposure assumptions and decisions within the biological evaluation plan and risk assessment records, thereby supporting regulatory compliance against ISO 10993-1 version 2025 and the selection of suitable testing protocols.
Conclusions
ISO 10993-1:2025 represents a significant evolution in the approach to biological evaluation of medical devices, emphasizing a risk-based, science-driven framework that considers both the nature and duration of device contact, potential chemical exposures, and patient-specific factors. The updated standard provides clearer guidance on categorizing devices, assessing reasonably foreseeable misuse, and integrating post-market data into ongoing risk management, ensuring that manufacturers can make informed decisions about biocompatibility testing while maintaining regulatory compliance. By focusing on total exposure, material characteristics, and potential bioaccumulation, ISO 10993-1:2025 helps align testing strategies with real-world use scenarios, ultimately enhancing patient safety and supporting more robust, evidence-based evaluations of medical device safety. This standard encourages proactive documentation, thorough planning, and continuous assessment, setting a new benchmark for consistency, transparency, and scientific rigor in medical device evaluation worldwide.






One Comment
Comments are closed.