Hidden Rules of 510k: What FDA Really Expects
The 510k process is the most common pathway for bringing medical devices to market in the United States. Named after Section 510(k) of the Federal Food, Drug, and Cosmetic Act, it requires manufacturers to demonstrate that their device is substantially equivalent to a legally marketed predicate device. The process is designed to ensure safety and effectiveness without the need for full premarket approval (PMA), making it faster and less costly for many devices.
Table of Contents
Type of 510k Submissions
The FDA recognizes several types of 510k submissions, each designed to accommodate different scenarios for demonstrating substantial equivalence to a legally marketed predicate device. Understanding the distinctions between them is critical to selecting the most appropriate pathway and ensuring a smooth review process.
Traditional 510k
The traditional 510k submission is the most used for the majority of the medical devices. It requires a complete compilation of technical documentation, labeling, performance data, and any other supporting evidence needed to demonstrate substantial equivalence. This type of submission is suitable for new devices or when changes to an existing device are significantly enough that other streamlined submission types are not appropriate.
Abbreviated 510k
The abbreviated 510k relies heavily on FDA-recognized consensus standards, guidance documents or special controls. The goal is to reduce the burden of repetitive testing by demonstrating conformity to established standards; it is a submission particularly useful for devices that closely follow existing regulatory guidance and have well-characterized technologies.
Special 510k
The Special 510(k) submission is intended for modifications to a device that already has 510(k) clearance. It allows for a faster review because the FDA is already familiar with the original device, and it is applicable when the changes do not affect the device’s intended use or alter its fundamental technology in a way that could raise new safety or effectiveness concerns.
The Concept of Substantial Equivalence
The concept of substantial equivalence (SE) is central to the 510k regulatory pathway for medical devices in the United States and serves as the foundation upon which the FDA evaluates whether a new device can be legally marketed without undergoing the more rigorous Premarket Approval (PMA) process.
Substantial equivalence does not require that the new device be identical to a previously cleared predicate device; rather, it requires that the device demonstrate safety and effectiveness comparable to that of an already legally marketed device, considering both the intended use and technological characteristics.
In practice, SE is determined through a thorough comparison that examines the intended use, indications for use, design features, materials, energy sources, and performance characteristics of the new device in relation to the predicate. The concept recognizes that innovation and minor technological improvements are permissible, provided that they do not introduce new risks or alter the fundamental purpose of the device.
The FDA also considers whether any differences between the new device and its predicate could impact safety or effectiveness, requiring manufacturers to provide justification, risk analyses, or additional testing to demonstrate equivalence. This concept ensures that new devices can enter the market efficiently while maintaining a regulatory safeguard, encouraging innovation within a framework of established safety standards.
Do you Need Clinical Data for a 510k process?
The assessment of the need for clinical data shall be performed prior to initiating the preparation of a 510(k) submission. Typically, clinical data (intended as data derived from clinical investigations conducted on human subjects) are not required for most 510k submissions. However, there are circumstances in which the 510(k) process may need to include data from clinical investigations, such as:
- When the predicate device included clinical data to support its clearance;
- When there are differences between the subject device and the predicate device that cannot be adequately justified or substantiated without clinical data;
- When it is not possible to demonstrate substantial equivalence to the predicate device without a clinical investigation;
- When clinical data are explicitly requested by the FDA.
A particular consideration applies to Software as a Medical Device (SaMD). In most cases, clinical data are not required for these devices, as they are typically intended to collect, process, or analyze data rather than to directly perform a therapeutic intervention. Nevertheless, the FDA requires adequate performance evaluation of the device, which often involves testing the software using real clinical data to demonstrate its accuracy, reliability, and intended functionality. In such cases, two main approaches may be considered:
- A formal clinical investigation is conducted, similarly to other types of medical devices;
- Existing clinical data are obtained and used to demonstrate the performance of the device, without conducting a new clinical investigation.
Does FDA check my QMS for a 510k?
The 510(k) process is intended to obtain clearance to market a medical device in the United States. All Class II medical devices must obtain 510(k) clearance before being marketed in the United States, unless they are specifically exempt from this requirement.
During the review process, the FDA focuses on the technical documentation of the device and does not assess the manufacturer’s quality management system (QMS) documentation. However, it is important to note that, in order to market medical devices in the United States, compliance with QMS requirements—particularly the Quality Management System Regulation (QMSR) and ISO 13485—is mandatory. In other words, although the FDA does not review QMS documentation as part of the 510(k) process, manufacturers are still required to be fully compliant with applicable QMS requirements at all times.
AUDIT-READY KIT
Build your ISO 13485 QMS with confidence.
Built on 15+ years of audit experience — every SOP and template references the regulations auditors expect. Get to certification faster, with industry best practices baked in.
- ✓30 SOPs covering the full QMS scope
- ✓56 templates ready to customize
- ✓Aligned with EU MDR + FDA QMSR
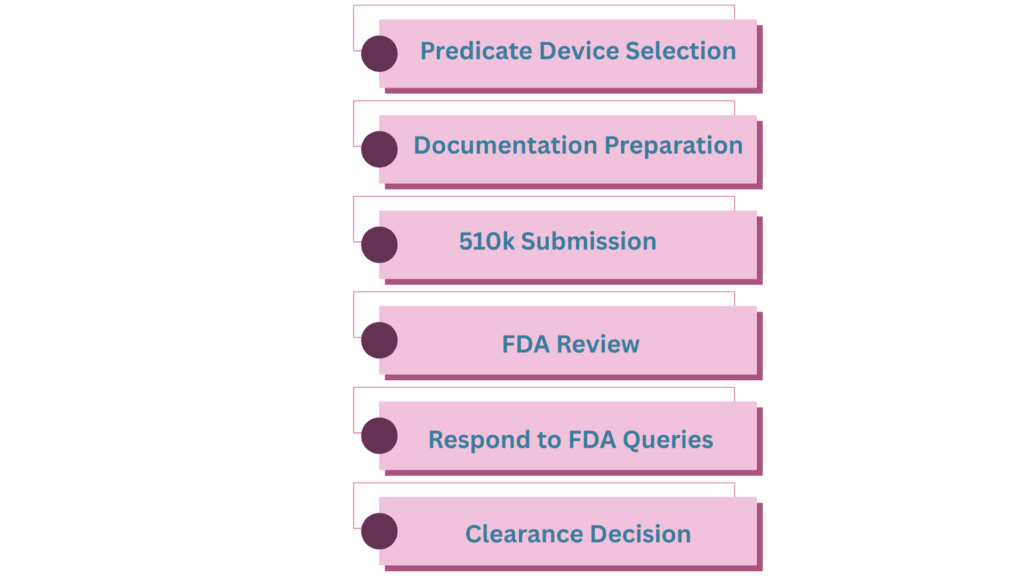
510k Submission in Practice
The 510(k) submission is prepared using a specific interactive template known as the eSTAR (electronic Submission Template And Resource). This template is provided in PDF format and includes a structured series of questions designed to guide the applicant through the preparation of a complete and compliant submission. Within the eSTAR template, applicants are required to provide detailed information about the device and upload supporting documentation related to its technical, non-clinical, and regulatory characteristics.
The eSTAR template is available for download from the FDA website and is designed to streamline the submission process while ensuring consistency and completeness. It contains multiple sections that may dynamically appear or be omitted depending on the specific characteristics of the device, such as its classification, intended use, technological features, and regulatory requirements. This adaptive structure helps ensure that only relevant information is requested, reducing unnecessary burden while maintaining regulatory rigor.
For example, in the case of a single-use, non-active medical device, the main sections typically included in the submission are:
- Cover Letter and Applicant Information
- Device Description
- Indications for Use
- Predicate Devices and Substantial Equivalence
- Labeling
- Reprocessing, Sterility, and Shelf-Life
- Biocompatibility
- Performance Testing
- Administrative Documentation
Each of these sections requires the applicant to provide specific information and supporting evidence to demonstrate that the device is safe, performs as intended, and is substantially equivalent to a legally marketed predicate device. Proper completion of the eSTAR template is essential to facilitate an efficient FDA review and to minimize the likelihood of requests for additional information.
The Biocompatibility section in particular must present the biological evaluation for every material that contacts the body, built per the FDA-modified ISO 10993-1 matrix — including chemical characterization and, where required, in vivo sensitization data. See our guide to biocompatibility testing requirements for what FDA expects here.
Can a device be Exempted from 510k Submission?
Certain medical devices may be exempt from the requirement to submit a 510k notification, depending on their classification and intended use. Generally, many Class I devices and a limited number of Class II devices are exempt from 510(k) submission, provided they are considered low risk and do not raise significant safety or effectiveness concerns. However, even when a device is exempt, manufacturers must still comply with applicable regulatory requirements, including registration and listing, labeling regulations, and quality management system requirements.
FDA has published a specific list of exempted medical devices, which are available at this link. As it can be seen, some class devices are exempted , for example:
- Some Neurological Devices
- Some Dental Devices
- Some Anesthesiology Devices
- and many more
It is also important to note that exemption may not apply if the device has novel features, new intended uses, or deviates significantly from existing exempted device types, in which case a 510(k) submission may still be required.
What Happens if It is not Possible to Demonstrate Substantial Equivalence
If it is not possible to demonstrate substantial equivalence to a legally marketed predicate device, the FDA will issue a determination of “Not Substantially Equivalent” (NSE). In such cases, the device cannot be cleared through the 510k pathway and cannot be legally marketed in the United States under this route.
In these circumstances, the manufacturer must consider alternative regulatory pathways, most commonly the De Novo classification process or, for higher-risk devices, the Premarket Approval (PMA) pathway.
The De Novo process may be appropriate if the device is considered low to moderate risk but lacks a suitable predicate, allowing it to be classified into Class I or II and potentially serve as a future predicate for similar devices. Conversely, if the device presents higher risks or insufficient evidence of safety and effectiveness, a PMA application—requiring more extensive data, often including clinical evidence—may be necessary. Therefore, the inability to demonstrate substantial equivalence significantly impacts regulatory strategy, timelines, and costs, and highlights the importance of careful predicate selection and early regulatory planning.
510k Fee & Small Business License Number
When submitting a 510k notification to the FDA, applicants are required to pay a user fee unless they qualify for a reduced fee under the Small Business Program. T
he 510k fee varies annually and depends on whether the submitter qualifies as a small business. To be eligible for the reduced fee, a company must obtain a Small Business Determination (SBD) Number from the FDA, which confirms the company meets the criteria for small business status based on revenue and other factors. This number must be included on the 510(k) submission to claim the fee reduction.
It is important to note that the SBD Number must be obtained before submitting the 510k, as failure to include it can result in the full fee being charged and may delay the review process. This program allows startups and smaller manufacturers to bring devices to market while reducing financial barriers. The Small Business Licence Number is issued on a yearly basis and it must be renew every year.
To obtain a small business license number, the manufacturer must submit the FDA Form 3602, which must then be submitted to the agency through the specific FDA portal. Once the FDA reviews the submitted documentation, the small business license number is issued and communicated to the manufacturer.
Regulatory Compliance for 510k Submissions: FDA Consensus Standards
In the framework of 510(k) submissions, adherence to FDA-recognized consensus standards plays a critical role in demonstrating regulatory compliance and supporting the safety and effectiveness of a medical device. These standards, which are developed by recognized international or national standards organizations such as ISO, IEC, or ASTM, provide well-established methods for testing, performance evaluation, biocompatibility, electrical safety, software validation, and other device-specific requirements.
By referencing these standards in a 510(k) submission, manufacturers can streamline the review process, as compliance with recognized standards can serve as objective evidence of conformity with FDA requirements. In many cases, using consensus standards reduces the need for extensive additional testing or clinical data, particularly for well-characterized device types.
The FDA maintains an up-to-date list of recognized standards, and the eSTAR template for 510(k) submissions includes sections to indicate which standards have been applied. Importantly, when a manufacturer claims conformance to a standard, they must provide a statement of conformity and include test reports or evidence demonstrating compliance. Overall, proper use of FDA-recognized consensus standards strengthens the submission, increases predictability of review outcomes, and ensures that the device meets widely accepted safety and performance benchmarks.
MULTI-MARKET KIT
One audit. Five markets. Ready to submit.
Our MDSAP Documentation Kit covers Brazil ANVISA, Japan PMDA, Canada Health Canada, Australia TGA, and FDA — with country-specific reportability worksheets and application checklists you can use today.
- ✓15 SOPs covering 5 MDSAP markets
- ✓18 templates with country worksheets
- ✓Brazil · Japan · Canada · Australia · USA
Conclusions
The 510(k) pathway is a cornerstone of medical device regulation in the United States, providing a structured and efficient mechanism for demonstrating that a device is safe, effective, and substantially equivalent to a legally marketed predicate device. Understanding the different submission types—Traditional, Abbreviated, Special, and Third-Party 510(k)—is essential for selecting the most appropriate pathway based on device classification, technological characteristics, and any modifications to existing products.
Critical aspects of a successful submission include careful predicate selection, thorough technical documentation, proper use of FDA-recognized consensus standards, and a clear assessment of the need for clinical data. While the FDA primarily reviews the technical evidence, compliance with Quality Management System requirements, including ISO 13485 and the QMS Regulation, remains mandatory for all manufacturers.
Additional considerations, such as 510(k) fees and eligibility for small business reductions, must also be addressed early in the process to avoid delays. By strategically planning the 510(k) submission, adhering to regulatory guidance, and providing complete and well-documented evidence, manufacturers can facilitate a smooth review process, minimize the risk of “Not Substantially Equivalent” determinations, and ultimately bring safe and effective medical devices to market efficiently.