ISO 13485 Supplier Management: Qualification, Approved Supplier List and Audits
SO 13485 supplier management is one of the most operationally complex requirements of the standard — and one of the most consistently found-deficient in Notified Body audits and FDA inspections. Supplier management is a regulatory requirement and a critical quality system element audited by Notified Bodies and regulatory authorities. Core elements include supplier qualification, risk classification, quality agreements, performance monitoring, audit scheduling, nonconformance handling, and supplier re-evaluation. Supplier records must demonstrate traceability from supplier selection to CAPA or requalification decisions.
Every component, material, and service that enters a medical device carries with it the quality and regulatory compliance of the supplier who provided it. A device that meets all internal quality requirements but incorporates a component from an unqualified or poorly controlled supplier is not a compliant device — and regulators will hold the manufacturer responsible regardless of where the fault originated.
Purchasing controls and supplier oversight are recurring pain points in FDA inspections, and Clause 820.50 continues to show up as a frequently cited area. Under QMSR 2026, “approved” must mean a controlled status backed by documented criteria, evaluation evidence, purchasing requirements flowed down, and ongoing monitoring tied to what that supplier actually provides.
This guide covers the complete ISO 13485 Clause 7.4 supplier management framework — from risk-based classification through qualification, the Approved Supplier List, purchasing controls, supplier audits, and the significant changes introduced by FDA QMSR in February 2026. For context on the broader ISO 13485 quality management system within which supplier management operates, see our complete ISO 13485 guide. For information on how supplier audit findings integrate with the corrective action process, see our ISO 13485 CAPA procedure guide.
AUDIT-READY KIT
Build your ISO 13485 QMS with confidence.
Built on 15+ years of audit experience — every SOP and template references the regulations auditors expect. Get to certification faster, with industry best practices baked in.
- ✓30 SOPs covering the full QMS scope
- ✓56 templates ready to customize
- ✓Aligned with EU MDR + FDA QMSR
What ISO 13485 Clause 7.4 Requires
ISO 13485:2016 addresses supplier management in Clause 7.4 (Purchasing) with three sub-clauses: 7.4.1 (Purchasing Process), 7.4.2 (Purchasing Information), and 7.4.3 (Verification of Purchased Product). Additional requirements for outsourced processes appear in Clause 4.1.5.
ISO 13485:2016 Clause 7.4 requires that supplier evaluation and control be proportionate to the risk associated with the medical device. Additionally, supplier audit records are now subject to FDA inspection, changing the documentation standards for supplier qualification activities.
The primary supplier management requirements under ISO 13485:2016 include: Supplier Evaluation and Selection (Clause 7.4.1) — manufacturers must establish and document criteria for evaluating and selecting suppliers based on their ability to meet specified requirements and the associated risk; Supplier Monitoring and Re-evaluation (Clause 7.4.1) — monitoring and re-evaluation must be conducted at defined intervals or upon significant changes; and Quality Agreements (Clauses 4.1.5 and 7.4.2) — quality agreements must clearly define product specifications, quality requirements, change control requirements, and regulatory responsibilities.
The three pillars of the framework are: evaluation and selection before first purchase, purchasing controls through documented information, and verification and ongoing monitoring throughout the entire supply relationship.
Risk-Based Supplier Classification
The most critical principle in ISO 13485 supplier management is proportionality. The extent of qualification, monitoring, and control must match the risk that the supplied product or service poses to device safety, performance, and regulatory compliance.
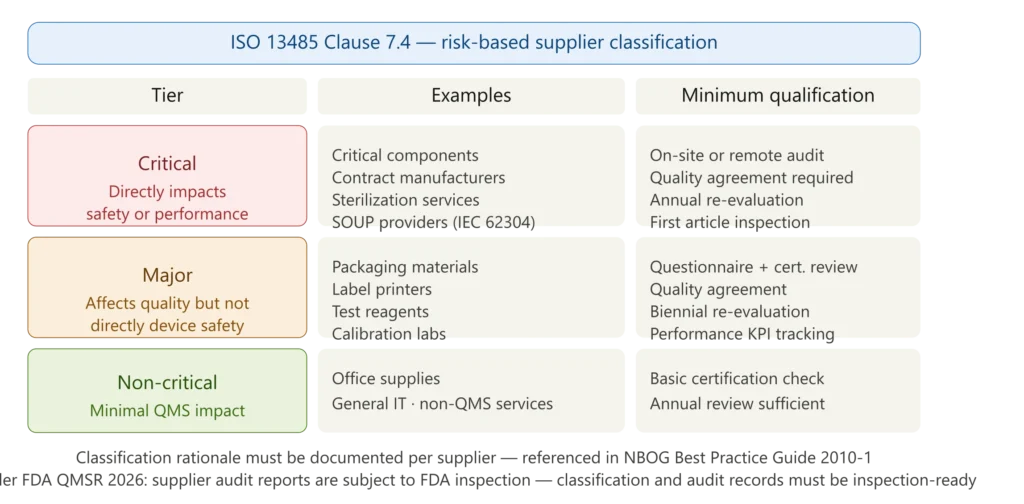
Figure 1 — ISO 13485 supplier risk classification framework
One effective practice is linking supplier qualification directly to product risk. For example, mapping suppliers against a simple risk scale — critical, important, or general — where critical suppliers require on-site audits, important ones go through detailed questionnaires, and general suppliers only need basic checks. It keeps the process lean while still satisfying auditors.
The classification must be documented with explicit rationale for each supplier. For software providers, the risk classification must also consider the SOUP (Software of Unknown Provenance) requirements of IEC 62304 — SOUP providers are a category of critical suppliers whose products require qualification, version control, anomaly monitoring, and risk assessment per our SOUP management guide.
The Supplier Qualification Process
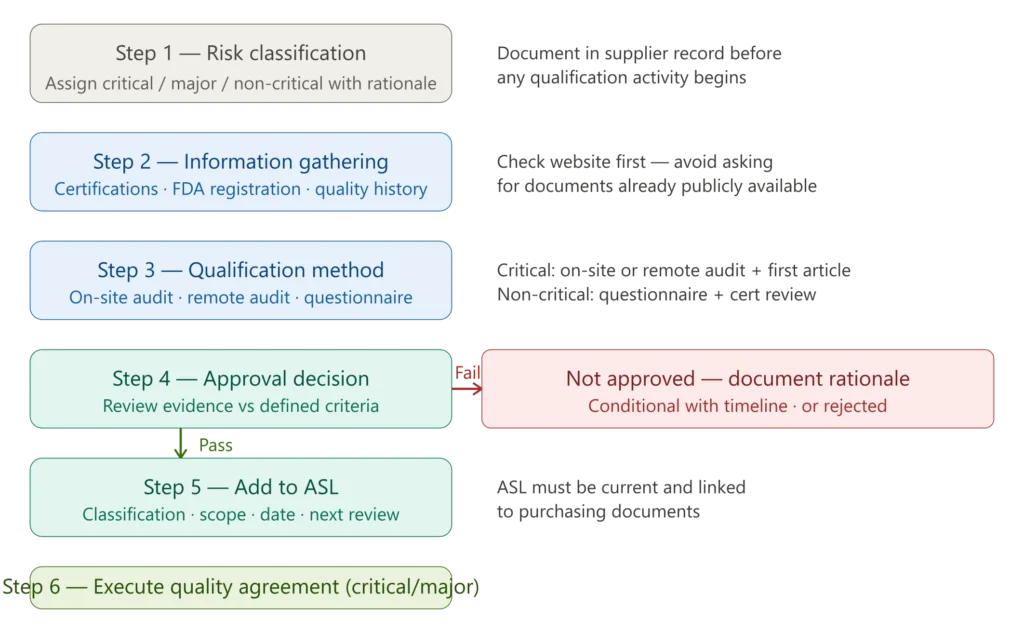
Figure 2 — ISO 13485 supplier qualification process flowchart
The goal of the supplier evaluation is to determine if the supplier meets your requirements and can be added to the Approved Supplier List. Nothing related to final product quality should be purchased from suppliers that are not on the ASL — this puts manufacturers at serious risk of bad quality products, audit findings, and regulatory consequences. EU Artificial Intelligence Act
Step 1 — Risk classification: Determine whether the supplier is critical, major, or non-critical based on the impact of their product or service on device safety and performance. Document the classification rationale before any qualification activity begins.
Step 2 — Information gathering: Collect objective evidence of supplier capability — quality certifications (ISO 13485, ISO 9001), FDA registration where applicable, regulatory compliance history, and delivery performance data. Check the supplier’s website for certifications before sending out requests — if a supplier has a current ISO 13485 certification easily available, it shows a lack of empathy to ask for it by email. EU Artificial Intelligence Act
Step 3 — Qualification method selection: The method must be proportionate to the supplier’s risk classification. If the supplier is high-risk, schedule or perform the supplier audit. If they are providing a product, request a first article to ensure it meets specifications. Transcend
Step 4 — Approval decision: Review all collected evidence against defined qualification criteria. Document the outcome — approved, conditionally approved with a defined timeline, or not approved — with full rationale.
Step 5 — Add to the Approved Supplier List: Add approved suppliers with their classification, scope of approval, approval date, and next re-evaluation date.
Step 6 — Execute the quality agreement: For critical and major suppliers, execute a formal quality agreement before the first purchase.
COMPLETE CATALOG
Find the documentation you need — instantly.
Whether you need a complete kit or just one specific SOP, our catalog has it. 45 process packages and 3 complete bundles, all instantly downloadable and fully editable.
- ✓Complete bundles or individual packages
- ✓45 process packages from €69 each
- ✓ISO 13485 · MDSAP · Combined Kit
The Approved Supplier List (ASL) — Clause 7.4.1
The backbone of the supplier management system is records. At minimum, you should be able to show: an Approved Supplier List that is kept current, supplier evaluations and approval records, and evidence of monitoring including complaints, scorecards, and audit results.
A compliant ASL must include for each supplier: supplier name and address, risk classification (critical/major/non-critical), scope of approval specifying exactly which products or services are approved, approval date, qualification status, next re-evaluation due date, and key quality contact. The scope of approval is particularly important — a supplier approved for one product or process is not automatically approved for a different product or process. Under QMSR, FDA has stated it can inspect supplier audit reports along with management review and quality audit records. So “approved” must become a status that can be defended with objective evidence, not institutional memory.
Purchasing must be prevented from using non-listed suppliers for quality-relevant items. The ASL is a purchasing control, not just a record — and the link between the ASL and active purchase orders must be demonstrable on demand.
Purchasing Controls — Clause 7.4.2
Purchasing information must precisely specify what is being purchased and the quality requirements that apply. Quality agreements must clearly define product specifications, quality requirements, change control requirements, and regulatory responsibilities. A specific ISO 13485 requirement that is frequently overlooked is the change notification agreement: purchasing information shall include, as applicable, a written agreement that the supplier notify the organization of changes in the purchased product prior to implementation of any changes that affect the ability of the purchased product to meet specified purchase requirements.
Every purchase order for a quality-relevant item must reference the product specification at a specific revision level — meaning the supplier cannot ship against an outdated specification without the manufacturer’s knowledge. Purchase orders tied only to part numbers without version control are a consistent audit finding under both ISO 13485 and FDA QMSR.
Supplier Audits — When and How
While it is not required by ISO 13485 and the FDA does not specify in the CFR that you must audit suppliers, it is a very good idea to audit your critical suppliers. If an auditor or FDA inspector sees evidence that your current purchasing controls are inadequate, performing supplier audits may be forced as a corrective action.
Under QMSR, FDA will now frequently request supplier audit reports — a practice virtually unheard of before. Every audited company should be ready to furnish those records. This essentially raises the bar on every quality agreement and supplier evaluation, since the audit findings themselves could become inspection findings.
Common audit problems and solutions include: audit reports that identify issues but show no corrective action follow-up — track all findings to closure with evidence before closing; auditor qualification not documented — maintain auditor training records showing ISO 19011 or equivalent training; audit scope not matching supplier risk — document scope rationale based on supplier classification and products supplied; and findings without clear requirement references — each finding should cite the specific requirement (ISO clause, specification section, or quality agreement provision) that was not met.
Supplier audit findings must feed directly into the ISO 13485 CAPA procedure — they are a formal trigger for corrective action and must be tracked to closure with effectiveness verification before the CAPA can be closed. For manufacturers pursuing MDSAP certification, supplier management is assessed across all five participating regulatory authorities under MDSAP Chapter 7.
FDA QMSR 2026 — What Changed for Supplier Management
Under QSR, 21 CFR 820.180(c) explicitly exempted supplier audit records from FDA inspection. Under QMSR, 820.180(c) has been revised to remove this exemption, and QSIT will be officially withdrawn February 2, 2026.
Critical supplier management under QMSR — suppliers involved in high-risk areas such as sterilization, software, and component manufacturing — need more thorough audits, validation evidence, and continuous performance monitoring. OEMs cannot delegate accountability. The legal manufacturer holds full responsibility for product quality. Supplier risks should be reported to management review, with documented actions taken.
The practical implications for manufacturers are: supplier audit reports must now be audit-ready for FDA inspection with complete, specific objective evidence; the risk-based classification approach must be documented with explicit rationale; and ongoing re-evaluation records must demonstrate that periodic monitoring was actually conducted, not just scheduled.
✦ PREMIUM BUNDLE
The ultimate global QMS documentation bundle.
Combine ISO 13485 + all 5 MDSAP markets in one premium package. Deduplicated structure means you customize each document once — not twice. Save €199 vs buying the kits separately.
- ✓41 SOPs covering both ISO 13485 and MDSAP
- ✓70+ templates with deduplicated structure
- ✓Save €199 vs buying separately
FDA QMSR 2026 — What Changed for Supplier Management

Figure 3 — Approved Supplier List mandatory content
Ongoing Supplier Monitoring and Re-evaluation
ISO 13485 requires that manufacturers continuously monitor and verify the performance of their suppliers. Regular reviews of supplier performance should cover quality, delivery reliability, and compliance with contractual agreements. Manufacturers should periodically audit suppliers to assess their operations and verify that they are adhering to agreed-upon quality and regulatory standards. Orrick
Performance monitoring KPIs should be defined in the supplier management procedure and tracked for each critical and major supplier. Common KPIs include: incoming inspection rejection rate, on-time delivery rate, certificate of conformance timeliness, supplier-initiated change notification compliance, and SCAR response time.
Re-evaluation triggers beyond the scheduled periodic review include: a significant change to the supplier’s quality system, manufacturing process, or key personnel; a product nonconformance traceable to the supplier; or a regulatory action involving the supplier. Supplier Corrective Action Requests (SCARs) are the formal mechanism for addressing supplier nonconformances — and they must be tracked through the CAPA system with documented effectiveness verification before closure.

Figure 4 — Most common ISO 13485 supplier management audit findings
Frequently Asked Questions
Is an Approved Supplier List explicitly required by ISO 13485? Maintaining an Approved Supplier List is not explicitly named in the standard text, but it is the universally expected practical implementation of Clause 7.4.1’s requirement that suppliers be evaluated and selected based on documented criteria. Every Notified Body and FDA inspector expects to see an ASL during QMS audits — its absence is an immediate finding.
How often must suppliers be re-evaluated under ISO 13485? Re-evaluation must occur at defined intervals specified in your supplier management procedure, based on supplier risk classification. Critical suppliers are typically re-evaluated annually, major suppliers biennially. Re-evaluation must be documented with objective evidence of activities actually conducted.
Are quality agreements required for all suppliers? Quality agreements are required for outsourced processes under Clause 4.1.5 and strongly expected for critical suppliers. For major suppliers, a quality agreement — or at minimum a change notification agreement embedded in the purchase order terms — is expected under Clause 7.4.2. Non-critical suppliers typically require only standard terms and conditions covering specification compliance.
Do supplier audit reports have to be shared with the FDA under QMSR? Yes — under QMSR, 820.180(c) has been revised to remove the old inspection exemption for supplier audit records. FDA inspectors can now request supplier audit reports as part of a QMSR inspection. All supplier audit records must be complete, traceable, and maintained to the same standard as other QMS records.
Conclusions
ISO 13485 supplier management is not a peripheral compliance activity — it is a foundational element of device quality that directly affects patient safety, regulatory compliance, and audit outcomes. Companies who build strong supplier management processes don’t just pass audits more smoothly — they avoid costly surprises like recalls, production delays, and regulatory findings.
Under FDA QMSR 2026, the bar for “approved supplier” has been definitively raised. “Approved” must now be a controlled status backed by documented criteria, evaluation evidence, and ongoing monitoring — not a static entry in a spreadsheet that was last reviewed years ago.
The ISO 13485 Supplier Management Documentation Package on MD Regulatory includes a complete supplier management procedure aligned with Clause 7.4, an Approved Supplier List template, supplier qualification form, supplier audit checklist aligned with ISO 13485 internal audit standards, quality agreement template, and SCAR form — all immediately deployable in an existing ISO 13485 QMS.
Related articles: ISO 13485 Complete Guide · ISO 13485 Internal Audit Checklist · ISO 13485 CAPA Procedure · SOUP Management IEC 62304 · MDSAP Audits