FDA De Novo Pathway: When to Use It and How to Submit
The FDA De Novo pathway is one of the most strategically important — and most misunderstood — routes to US market authorization for medical devices. It exists to solve a specific regulatory problem: what happens when you develop a novel, low-to-moderate-risk device that has no legally marketed predicate device to compare against?
The De Novo pathway provides marketing authorization for novel, low-to-moderate-risk devices without a predicate, creating a new Class I or II classification with general or special controls. It is authorized by a grant order, not a clearance — and it creates a product code that future similar devices can reference through the 510(k) pathway. De Novo sponsors essentially create the regulatory roadmap that competitors will follow, providing a significant first-mover advantage.
For manufacturers of digital health tools, AI-powered diagnostics, novel wearables, and innovative combination technologies, understanding when and how to use the De Novo pathway is a critical regulatory strategy decision. This guide covers the complete FDA De Novo framework updated to 2026, including the eSTAR mandatory submission requirement introduced in October 2025. For context on how De Novo fits within FDA pathway selection for software products, our guide on Software as a Medical Device classification covers the full SaMD regulatory landscape.
COMPLETE CATALOG
Find the documentation you need — instantly.
Whether you need a complete kit or just one specific SOP, our catalog has it. 45 process packages and 3 complete bundles, all instantly downloadable and fully editable.
- ✓Complete bundles or individual packages
- ✓45 process packages from €69 each
- ✓ISO 13485 · MDSAP · Combined Kit
What Is the FDA De Novo Pathway?
Before 1997, any novel device without a predicate was automatically classified as Class III — requiring a full Premarket Approval (PMA) regardless of actual risk level. The De Novo Classification Request is the regulatory mechanism used when manufacturers have a novel medical device that is not high risk and should therefore be Class II or Class I. A De Novo is submitted when the manufacturer has determined there is no appropriate predicate device to claim substantial equivalence to — or after a 510(k) was submitted with a verdict of not substantially equivalent.
Once granted, the device type is granted a product code and a specific classification. Subsequent devices can refer to that device as a predicate device and can be found substantially equivalent to it through the 510(k) pathway.
De Novo vs 510(k) vs PMA — Choosing the Right Pathway
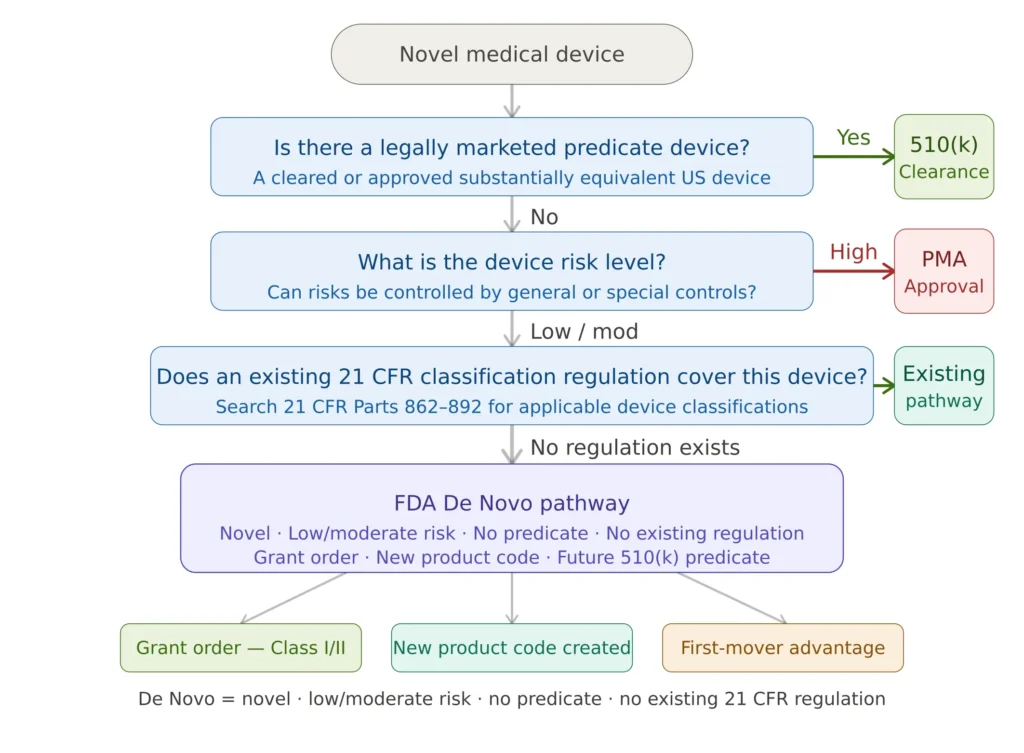
Figure 1 — FDA regulatory pathway decision flowchart
The three FDA pathways serve fundamentally different purposes. The 510(k) requires demonstration of substantial equivalence to a predicate — it is the right tool when an equivalent device exists. The PMA addresses high-risk Class III devices where general and special controls are insufficient. The De Novo fills the gap between them: novel devices of low-to-moderate risk with no predicate and no existing classification regulation.
User fees illustrate the cost difference clearly: a De Novo application costs $96,644 for standard companies and $24,161 for small businesses, compared to $10,953 and $2,738 respectively for a 510(k). PMA fees significantly exceed both. The De Novo investment is justified when the pathway creates a product code and classification that competitors must reference — making the De Novo grantee the regulatory standard setter for an entire device category.
When to Use the FDA De Novo Pathway
Use De Novo when all four conditions apply simultaneously: your device is novel with no legally marketed predicate; the device is low to moderate risk whose risks can be controlled by general or special controls; no existing classification regulation under 21 CFR Parts 862–892 covers your device type; and you can provide sufficient evidence to support the proposed Class I or II classification.
The De Novo pathway is ideal for digital health, diagnostics, and emerging tech. It requires robust risk mitigation and regulatory intelligence. The risk management system must demonstrate that risks are characterizable and mitigable — not simply asserted to be low.
Practical De Novo scenarios: A novel AI-powered clinical decision support tool with no existing predicate SaMD; a wearable biosensor measuring a physiological parameter in a way no cleared device does; a diagnostic test for a newly identified biomarker; a digital therapeutic with no existing cleared device in the same indication. For each of these, the benefit-risk analysis is a core submission requirement — demonstrating quantitatively that clinical benefits outweigh residual risks at the proposed classification level.
Of the 79 medical devices and IVDs cleared through the De Novo process between mid-2022 and mid-2024, all were given a Class II designation — reflecting the reality that truly low-risk Class I novel devices are rare. Most novel devices warrant De Novo have some degree of moderate risk requiring special controls.
De Novo Eligibility and Submission Requirements (21 CFR 860.220)

Figure 2 — De Novo submission content requirements
A De Novo request must comply with 21 CFR 860.220 and, from October 1, 2025, must be submitted through eSTAR. The FDA conducts an acceptance review that assesses whether the request meets administrative completeness requirements. Before listing the mandatory components, it is helpful to understand that FDA reviewers rely on clear evidence, structured reasoning and well-supported arguments for classification.
The proposed special controls section is among the most critical and most frequently deficient. Special controls must be specific, measurable, and directly linked to each identified risk — vague controls such as “performance testing shall be conducted” are routinely flagged as insufficient. The ISO 13485 quality management system documentation, including design controls and risk management evidence, forms a key part of the supporting package.
For SaMD devices, the performance data section must align with FDA’s software guidance and — where the device incorporates AI/ML — address the Predetermined Change Control Plan requirements covered in our SaMD classification guide.
eSTAR — The Mandatory Submission Format Since October 2025
Starting October 1, 2025, all De Novo submissions must be submitted as electronic submissions using eSTAR. The eSTAR is a dynamic PDF that includes logic and automation — it standardizes how information is provided, it doesn’t create new statutory requirements.
eSTAR is an interactive PDF form that guides applicants through the process of preparing a comprehensive medical device submission. By digitizing the submission process, eSTAR ensures that applicants provide all required information, improving the quality of submissions and reducing review time. The template aligns with regulatory requirements and includes automated cross-checks and prompts, helping manufacturers avoid common errors.
A common practical failure: teams finalize the technical content of their De Novo months in advance but never migrate it into the eSTAR template. When content is ported into eSTAR, the validator flags missing cross-references between indications and labelling — fixing one often reveals another. Large attachment files exceed size expectations, PDFs split, filenames change, links drift. Teams that discover this a week before target submission face serious delays.
The correct approach: start building the eSTAR file in parallel with preparing the technical content, not after. eSTAR’s built-in validation is a pre-submission quality check that eliminates many of the administrative deficiencies that trigger acceptance review failures.
✦ PREMIUM BUNDLE · ISO + MDSAP
The ultimate global QMS documentation bundle.
Combine ISO 13485 + all 5 MDSAP markets in one premium package. Deduplicated structure means you customize each document once — not twice. 122 files: 41 SOPs and 70+ templates with shared foundation.
- ✓122 files · 41 SOPs + 70+ templates, deduplicated
- ✓Save €199 vs buying the kits separately
The De Novo Review Process
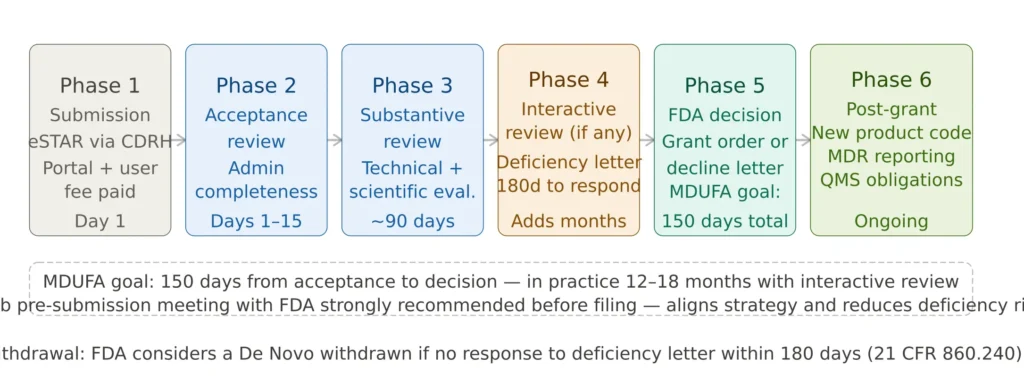
Figure 3 — FDA De Novo review timeline and process
Once a company determines its regulatory pathway, establishment registration and device listing are required for entities involved in US commercial distribution. Registration and listing allow the FDA to identify who makes or imports devices and which devices are in US commerce. These obligations apply after a De Novo grant, not before — but planning for them during submission preparation avoids post-grant delays in commercial launch. OpenRegulatory
The Q-Sub meeting is the single most cost-effective investment in De Novo preparation. Before committing to a full submission, a Pre-Submission meeting with the FDA allows manufacturers to confirm the De Novo is the right pathway, align on proposed special controls, and understand evidence expectations for the specific device. Skipping the Q-Sub to save time almost always costs more time in the long run.
Common De Novo Pitfalls
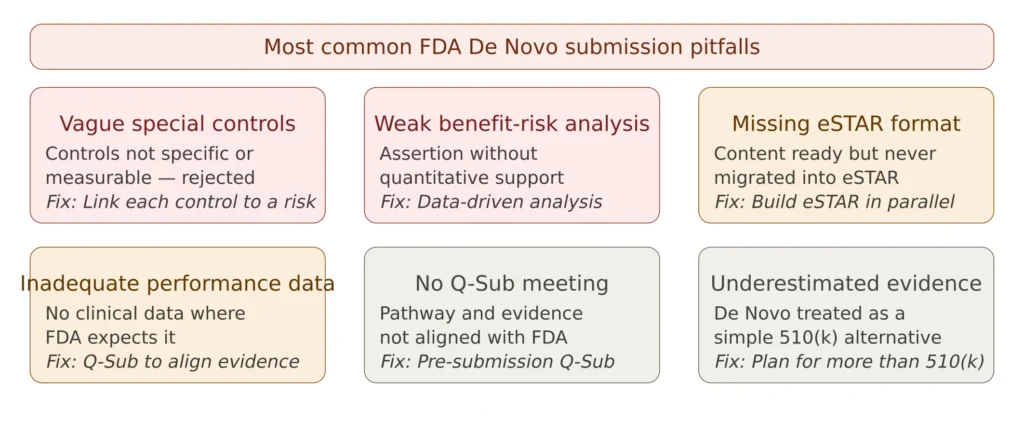
Figure 4 — Most common FDA De Novo submission failures
Vague special controls are the primary cause of De Novo deficiency letters. Controls must be specific, measurable, and directly linked to identified risks — not generic performance testing requirements. Insufficient benefit-risk analysis — the analysis must be data-driven and quantitative, not a narrative assertion. For guidance on structuring a defensible benefit-risk analysis, see our benefit-risk analysis guide.
Missing eSTAR format is a purely avoidable administrative failure. eSTAR is not new scientific requirements — it is a structured container that expects information to be complete and internally consistent. Teams that discover they have never built the eSTAR file a week before target submission face serious delays.
No Q-Sub meeting — the pre-submission meeting is the highest-return investment available before a De Novo submission. The FDA’s feedback on pathway appropriateness, evidence expectations, and proposed special controls eliminates the most costly surprises.
Frequently Asked Questions
How long does a De Novo review take? The FDA’s MDUFA performance goal is 150 days from acceptance to decision. In practice, interactive reviews with deficiency letters typically extend total review time to 12–18 months from initial submission to grant order. Complex novel technologies and submissions requiring clinical data review tend toward the longer end.
What is the FDA user fee for a De Novo in FY2026? FDA user fees for FY 2026 apply from October 1, 2025 through September 30, 2026. Small businesses qualified through the Small Business Determination program with gross receipts of $100 million or less are eligible for a reduced fee on De Novo requests. Check the FDA MDUFA fees page for the current FY 2026 fee schedule.
Is a De Novo the same as a 510(k) clearance? No. A De Novo is authorized by a grant order, not a clearance. While the practical market access result is similar, a De Novo creates an entirely new device classification, not a finding of substantial equivalence to an existing one.
Can a De Novo be resubmitted after decline? Yes — a declined De Novo can be resubmitted with additional data addressing the FDA’s concerns. The resubmission follows the same eSTAR process and requires a new user fee payment.
What happens to the 180-day withdrawal rule? FDA considers a De Novo request to have been withdrawn if the requester fails to provide a complete response to a request for additional information within 180 days after the date FDA issues such request. Timely, complete responses to AI letters are critical — partial or delayed responses restart the clock and can effectively terminate a submission.
Conclusions
The FDA De Novo pathway is the correct regulatory route for novel low-to-moderate-risk devices with no predicate — and when used strategically, it confers a first-mover advantage that no other FDA pathway provides. The investment in a thorough, well-prepared De Novo submission pays dividends not only in market authorization but in establishing the product code and classification standards that define an entire device category.
The three decisions that most consistently determine De Novo success are: engaging the FDA early through a Q-Sub meeting; investing in specific, risk-linked special controls; and building the eSTAR submission in parallel with technical content rather than as a last step.
This article is part of the MD Regulatory FDA series. Related articles: Software as a Medical Device (SaMD) Classification · Benefit-Risk Analysis under EU MDR · ISO 13485 Complete Guide · IEC 62304 Medical Device Software


One Comment
Comments are closed.