ISO 13485 CAPA Procedure: How to Manage Corrective and Preventive Actions
Table of Contents
The ISO 13485 CAPA procedure — covering Corrective and Preventive Actions — is consistently one of the most scrutinized elements of a medical device quality management system. In every Notified Body audit, every FDA inspection, and every MDSAP assessment, CAPA is examined in depth. And for good reason: a CAPA system that works is the clearest possible signal that an organization understands its own quality problems, fixes them at the root, and prevents them from recurring. A CAPA system that exists only on paper is, in regulatory terms, almost worse than no system at all.
From the regulatory point of view, the importance of CAPA cannot be understated. Device manufacturers are always evaluated on their CAPA processes during FDA inspections. The top reason for device manufacturers to receive FDA observations is due to CAPA and its related processes — and manufacturers also receive CAPA-related warning letters from FDA, mostly about quality system aspects concerned with CAPA.
This guide covers everything you need to build, operate, and document a compliant ISO 13485 CAPA procedure — from the regulatory requirements through the step-by-step process, root cause analysis methods, effectiveness verification, and the most common audit findings. If you are new to ISO 13485 and its quality management system requirements, we recommend reading our complete ISO 13485 guide first as the foundation for everything covered here.
What ISO 13485 Requires for CAPA
ISO 13485:2016 addresses CAPA in two separate clauses within Section 8.5 (Improvement):
Clause 8.5.2 — Corrective Action addresses nonconformities that have already occurred. The organization must take necessary actions to eliminate causes of nonconformities and prevent them from recurring. Any required corrective actions must be taken without any delay and should be of the same intensity as the nonconformities encountered.
Clause 8.5.3 — Preventive Action addresses potential nonconformities that have not yet occurred but have been identified as risks. The organization must determine actions needed to eliminate the causes of potential nonconformities to prevent their occurrence.
ISO 13485 divides CAPA into its two component concepts — corrective measures and preventive measures — addressed in Chapters 8.5.2 and 8.5.3 respectively. Despite separating these processes, both must be documented and evaluated to demonstrate improvement and preventive action, making CAPA the practical process by which both are united.
The standard requires a documented procedure covering all of the following elements for corrective action: reviewing nonconformities and determining their causes; evaluating the need for actions to ensure nonconformities do not recur; planning, documenting, and implementing required actions; updating documentation as a result of actions; verifying that corrective actions do not adversely affect the ability to meet applicable regulatory requirements or safety and performance requirements; and reviewing the effectiveness of corrective actions taken.
For preventive action, the same elements apply with the addition that preventive actions must be proportionate to the effects of potential problems — and that effectiveness verification is required “where appropriate.”
AUDIT-READY KIT
Build your ISO 13485 QMS with confidence.
Built on 15+ years of audit experience — every SOP and template references the regulations auditors expect. Get to certification faster, with industry best practices baked in.
- ✓30 SOPs covering the full QMS scope
- ✓56 templates ready to customize
- ✓Aligned with EU MDR + FDA QMSR
Corrective Action vs Correction vs Preventive Action — Getting the Terminology Right
One of the most persistent sources of confusion in CAPA management is the distinction between three related but distinct concepts:
Correction is the immediate action taken to address a specific nonconformity — fixing the defective product, recalling the batch, repairing the equipment. A correction addresses the symptom but not the cause.
Corrective action addresses the root cause of a nonconformity to prevent it from recurring. A corrective action follows from root cause analysis and targets the systemic failure that allowed the nonconformity to occur.
Preventive action addresses potential nonconformities — problems that have not yet occurred but have been identified through data analysis, risk assessment, trend monitoring, or process review as having a significant probability of occurring.
It is misleading to speak of a single “CAPA process.” Statements such as “The CAPA process begins with identifying the problem” demonstrate an inadequate understanding. Manufacturers generally need several processes to meet regulatory requirements — and combining corrective and preventive actions into one single undifferentiated process is just as imprecise as failing to distinguish between corrections and corrective actions.
In practice, most organizations implement a unified CAPA management system that handles both corrective and preventive actions through a common workflow — but with clearly differentiated triggers, investigation approaches, and documentation requirements for each type.
ISO 13485 CAPA Triggers — When to Open a CAPA
ISO 13485 CAPA Triggers — When to Open a CAPA
Not every quality event requires a CAPA. A proportionate, risk-based approach means applying CAPA to events of significance — where the underlying cause is systemic, where recurrence would pose a risk to product safety or regulatory compliance, or where the potential impact is significant.
Events that typically trigger corrective action:
- Internal audit nonconformities — for a detailed view of how audit findings feed into CAPA, see our ISO 13485 internal audit checklist guide
- Customer complaints indicating potential product safety or performance issues
- Nonconforming product findings during inspection or testing
- Process deviations with potential patient safety implications
- Adverse events or vigilance reports
- Supplier nonconformities with patient safety implications
- Regulatory inspection findings — including MDSAP audit findings
Events that typically trigger preventive action:
- Adverse trends identified in quality data (complaint rates, rejection rates, audit finding patterns)
- Risk assessment outputs identifying high-probability failure modes — directly connected to the benefit-risk analysisprocess required by EU MDR
- Industry-wide safety signals or regulatory guidance updates
- Management review outputs identifying systemic vulnerabilities
- Process hazard analyses identifying potential failure points
The decision to open a CAPA must be documented, including the rationale — both when a CAPA is opened and when a quality event is assessed and determined not to require one., including the rationale for the decision — both when a CAPA is opened and when a quality event is assessed and determined not to require one.
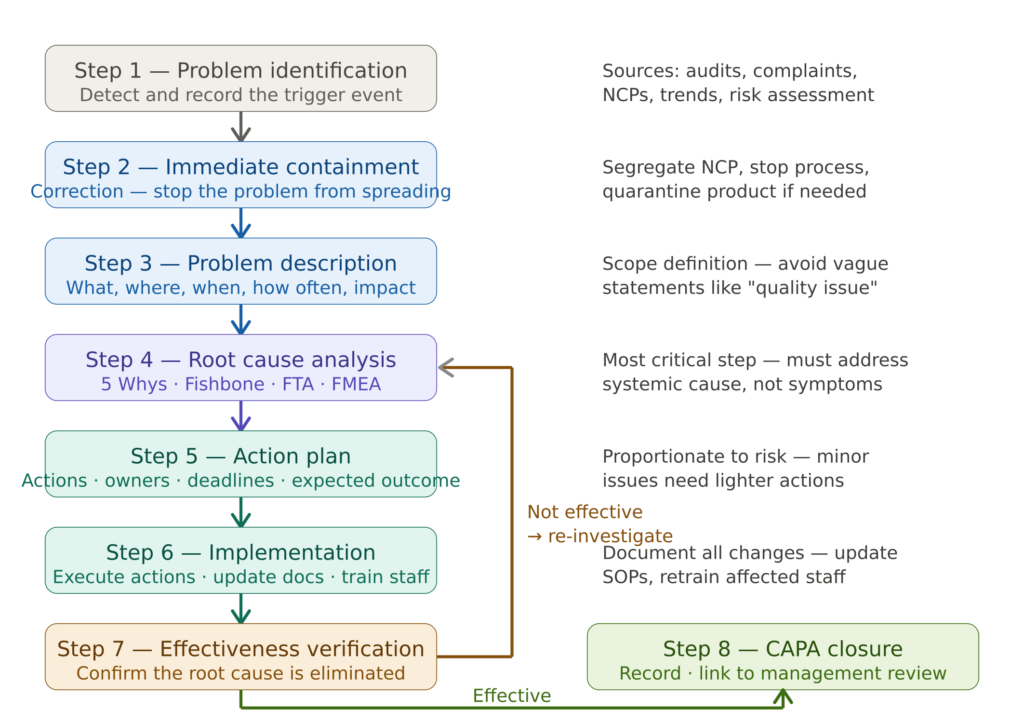
The ISO 13485 CAPA Process — Step by Step
Step 1 — Problem Identification and CAPA Initiation
The CAPA process begins with the identification of a trigger event and the formal opening of a CAPA record. The CAPA record must capture at minimum: the source of the trigger, the date of identification, the initial description of the problem, the person responsible for the investigation, and the assigned priority based on initial risk assessment.
Priority assignment is important — it determines how quickly the investigation must be completed and how quickly corrective actions must be implemented. A CAPA triggered by a potential patient safety issue requires faster response than one triggered by an administrative nonconformity.
Step 2 — Immediate Containment
Before investigating the root cause, the immediate impact of the nonconformity must be contained. This may involve segregating and quarantining nonconforming product, suspending a process, notifying customers or regulatory authorities, or issuing a field safety corrective action.
Containment is a correction — it addresses the specific instance of the problem. It must be documented and linked to the CAPA record, but it does not replace the corrective action that addresses the root cause.
Step 3 — Problem Description
A precise problem description is the foundation of an effective root cause investigation. It must answer five questions: What is the problem? Where was it observed? When was it first identified? How often does it occur? What is its potential impact on product safety, patient safety, or regulatory compliance?
A vague problem description — “supplier performance issue” — produces a vague investigation. A precise description — “28% of incoming inspection records for Component X from Supplier Y were missing the required signature in Field 4 during Q3 2025” — enables targeted root cause analysis.
Step 4 — Root Cause Analysis
Root cause analysis is the most critical and most frequently deficient step in CAPA management. If a CAPA skips root cause analysis or effectiveness checks, it becomes little more than a to-do list of corrections rather than a true improvement engine. Root cause work is where many CAPAs go wrong.
The root cause is the fundamental systemic reason why the problem occurred. “Operator error” is a symptom. “The work instruction was ambiguous and there was no independent verification required” is a root cause. Root cause analysis should always involve a cross-functional team — a single investigator will be limited by their own experience and perspective.
Step 5 — Action Planning
The action plan defines the specific corrective actions to be implemented, the owner responsible for each action, the target completion date, the expected outcome, and — critically — the effectiveness verification criteria. These criteria must be defined before implementation, not retrospectively.
Actions must be targeted at the root cause. A corrective action that retrains the operator without fixing the procedure that created the conditions for error will fail the effectiveness check — and generate the same finding in the next audit.
Step 6 — Implementation
All corrective actions must be implemented within the defined timelines. All changes must go through document control — updated procedures require formal revision, training on changes must be documented with evidence of completion and effectiveness evaluation. For significant changes affecting product safety or performance, a risk assessment of the proposed corrective action is required per ISO 13485 Clause 8.5.2(e).
This step directly connects to ISO 13485’s broader design control requirements — changes to manufacturing processes or device design triggered by CAPA must be evaluated against the full impact on the quality management system.
Step 7 — Effectiveness Verification
Effectiveness verification is the evidence-based confirmation that the corrective action eliminated the root cause and the problem has not recurred. The effectiveness check requires evidence-based review after a defined time or number of cycles to confirm the problem is controlled or risk reduced.
If effectiveness verification reveals the problem has recurred or the corrective action was insufficient, the CAPA is reopened and the root cause analysis revisited. This is not a failure — it is the system working as designed.
Step 8 — CAPA Closure and Management Review
A CAPA can be closed only when all actions have been completed, all documentation is updated, training has been conducted and documented, and effectiveness verification confirms resolution. CAPA trends must be reported at management review — giving top management visibility into systemic quality challenges.
MULTI-MARKET KIT
One audit. Five markets. Ready to submit.
Our MDSAP Documentation Kit covers Brazil ANVISA, Japan PMDA, Canada Health Canada, Australia TGA, and FDA — with country-specific reportability worksheets and application checklists you can use today.
- ✓15 SOPs covering 5 MDSAP markets
- ✓18 templates with country worksheets
- ✓Brazil · Japan · Canada · Australia · USA
Root Cause Analysis Methods for Medical Device CAPA
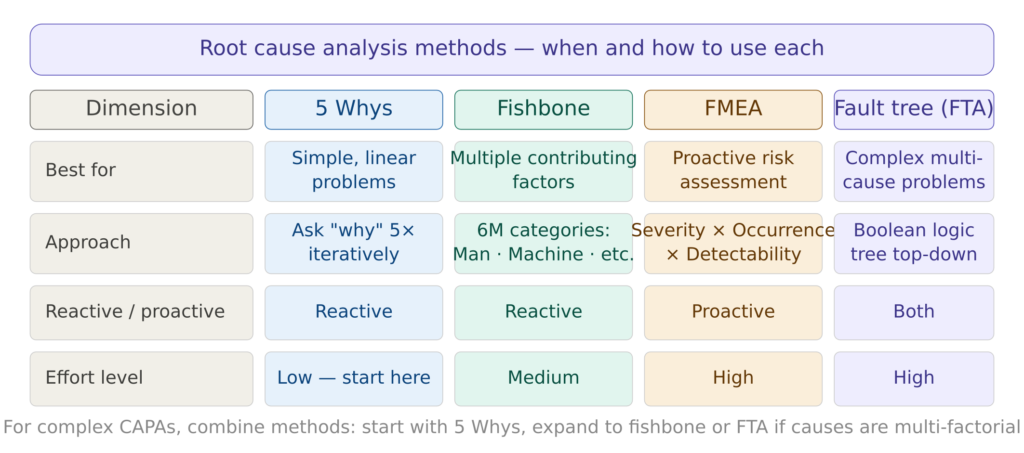
Figure 2 — Root cause analysis methods comparison
5 Whys is the most accessible root cause analysis method and the right starting point for most CAPAs. It involves asking “why” repeatedly until the systemic root cause is reached rather than stopping at a symptom. Most effective for linear, single-cause problems.
Fishbone diagram (Ishikawa) organizes potential causes into six categories — Man, Machine, Method, Material, Measurement, and Environment. It identifies contributing factors across these categories and is particularly effective when multiple factors may be contributing to the same problem. Works well in cross-functional team brainstorming sessions.
FMEA — Failure Mode and Effects Analysis is primarily a proactive tool used during design or process planning. FMEA assesses risks by scoring the severity, occurrence, and detectability of each failure mode, then prioritizes actions to mitigate them. In the CAPA context it is most useful for preventive actions. Note that FMEA is also central to risk management under ISO 14971 — our benefit-risk analysis guide covers how failure modes identified through FMEA feed into the broader risk management process.
Fault Tree Analysis (FTA) models cause-effect relationships graphically using Boolean logic, working top-down from the failure event to its root causes. Most appropriate for critical CAPAs with direct patient safety implications where multiple causes must combine for the failure to occur.
A combined approach utilizing multiple methods can be employed for more comprehensive analysis. A CAPA investigation might begin with the 5 Whys to identify immediate causes, followed by the fishbone diagram to categorize contributing factors, and finally fault tree analysis to map complex interactions between causes.
CAPA Documentation Requirements
ISO 13485 requires that records be maintained for all CAPA activities. A complete CAPA record must contain:
Problem description — the precise description of the nonconformity including source, date, frequency, scope, and initial risk assessment.
Immediate correction — what was done to address the specific instance of the problem.
Root cause analysis — the method used, the investigation process, the evidence reviewed, and the identified root cause.
Action plan — every action defined, with owner, target date, expected outcome, and effectiveness criteria.
Implementation evidence — documents, training records, validation data, or other objective evidence that actions were completed as planned.
Effectiveness verification — the criteria defined, the observation period, the data collected, and the conclusion.
Closure decision — who closed the CAPA, on what date, and based on what evidence.
Management review linkage — reference to the management review where CAPA status was reported.
For medical device software manufacturers, CAPA records related to software anomalies must also align with the anomaly resolution requirements of IEC 62304, which defines how software bugs discovered in post-production must be evaluated, tracked, and resolved within the QMS framework.

Featured Resource
CAPA SOP — ISO 13485, MDSAP & FDA QMSR
Audit-ready CAPA procedure + template + tracking database. Instantly downloadable in fully editable Word and Excel format.
CAPA and QMSR — What Changed in February 2026
The FDA’s Quality Management System Regulation (QMSR), effective February 2, 2026, incorporates ISO 13485:2016 by reference into 21 CFR Part 820. Under the old QSR, CAPA was a combined system that did not separate corrective versus preventive action in law. Under QMSR and ISO 13485, manufacturers must now maintain separate documented processes for corrective action (Clause 8.5.2) and preventive action (Clause 8.5.3), each with separate triggers and documentation requirements.
For organizations pursuing MDSAP certification, CAPA requirements are assessed across all five participating regulatory authorities — Australia, Brazil, Canada, Japan, and the FDA — making a fully documented, evidence-based CAPA system even more critical.

Figure 3 — Most common CAPA audit findings under ISO 13485
Symptom-level root cause is the single most common CAPA finding in FDA inspections. FDA inspectors often ask: “How do you ensure that your CAPA addresses the root cause rather than the symptom?” — the answer lies in consistent application of structured root cause analysis tools.
No effectiveness verification — CAPAs are closed once actions are implemented, without subsequent monitoring to confirm the problem did not recur. This is one of the clearest indicators of a CAPA system managed for compliance rather than improvement.
Inadequate timelines — CAPAs remain open for excessive periods with no documented progress or justification for delays. ISO 13485 requires corrective actions to be taken “without undue delay,” and Notified Bodies apply significant scrutiny to CAPA aging reports during surveillance audits. This is also one of the most visible findings during the ISO 13485 internal audit process.
Actions addressing symptoms rather than root causes — retraining the operator without fixing the underlying procedure. If the same training has been applied as a corrective action for the same type of error across multiple CAPAs, this pattern itself signals that root causes have never been properly identified.
Incomplete CAPA records — missing root cause analysis documentation, absent implementation evidence, or no formal closure record. Under FDA QMSR, CAPA records are now subject to FDA inspection.
No preventive action system — quality data is collected but never systematically analyzed to identify potential nonconformities and trigger preventive actions. Preventive action is consistently the most underused element of Clause 8.5 in most QMS implementations.
CAPA Effectiveness Verification — How to Do It Correctly
Effectiveness verification is the most frequently deficient element of CAPA management. The following principles define a robust approach.
Define criteria before implementation. Effectiveness criteria must be defined in the action plan — not decided retrospectively. Criteria should be measurable and specific: “zero recurrence of this nonconformity in the next 50 incoming inspection records” is measurable; “no further issues” is not.
Allow sufficient observation time. For a nonconformity that occurred twice in a quarter, a two-week observation period is insufficient. The observation period should encompass at least the same timeframe over which the original nonconformity was observed.
Use objective evidence. Effectiveness cannot be based on subjective assessment. It must be based on data, records, inspection results, or complaint rates.
Report results formally. The effectiveness verification must be documented in the CAPA record with the evidence reviewed, the conclusion, and the name and date of the person who verified effectiveness.
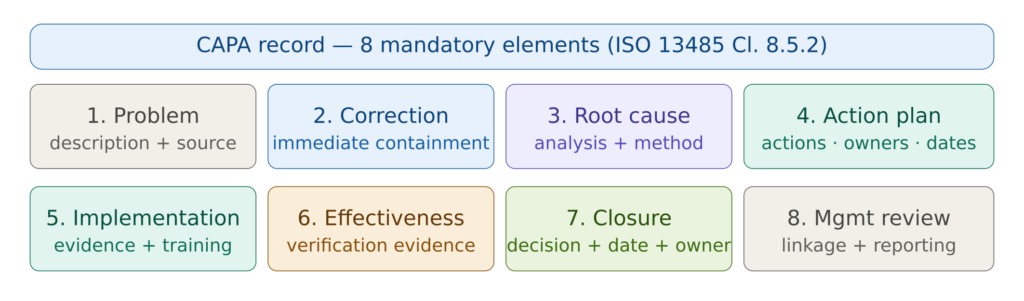
Figure 4 — CAPA record mandatory elements
All CAPA records must be retained for a minimum period defined in your document control procedure — typically the lifetime of the product plus the applicable regulatory retention period, with a general minimum of five years for most ISO 13485-certified organizations.
For manufacturers of medical device software, CAPA records triggered by software anomalies must be cross-referenced with the software anomaly resolution process defined under IEC 62304. The software anomaly list — which is also a key component of SOUP management for third-party components — feeds directly into the CAPA system when anomalies cross the threshold of regulatory significance.
Frequently Asked Questions
What is the difference between a correction and a corrective action? A correction addresses the specific instance of a nonconformity — fixing the defective product, correcting the erroneous record. A corrective action addresses the root cause to prevent recurrence. Both may be required for the same event but are distinct activities with different objectives and must be separately documented in the CAPA record.
How long should a CAPA remain open? ISO 13485 requires corrective actions to be taken “without undue delay.” There is no prescribed maximum duration, but most organizations define target timelines in their CAPA procedure — typically 30 to 90 days for most CAPAs, with shorter timelines for patient safety-related findings. Extended timelines must be formally justified and approved.
Is a CAPA required for every nonconformity? No. A proportionate, risk-based approach is appropriate. Minor, isolated nonconformities with no safety implications may be addressed through immediate correction without a formal CAPA. However, the decision not to open a CAPA must be documented and justified. If the same nonconformity recurs, a CAPA is required regardless of its severity — and recurring findings that were not CAPAed will themselves become a finding in the next internal audit.
How does CAPA connect to EU MDR post-market surveillance? EU MDR requires that post-market surveillance data feeds back into the risk management file and — where significant signals are identified — into the CAPA system. A PSUR identifying an adverse trend in complaint data or post-market performance should trigger a CAPA investigation. This integration between PMS and CAPA is one of the most closely examined connections in Notified Body surveillance audits, and connects directly to the benefit-risk analysis that must be maintained continuously throughout the device lifecycle.
Can preventive actions be triggered by positive data? Yes — and this is a sign of a mature QMS. Preventive actions can be triggered by risk assessments, regulatory guidance updates, process changes that introduce new failure modes, or management review decisions. Preventive action is not only reactive to negative trends — it is also a proactive management tool and one of the most underused elements of most QMS implementations.
COMPLETE CATALOG
Find the documentation you need — instantly.
Whether you need a complete kit or just one specific SOP, our catalog has it. 45 process packages and 3 complete bundles, all instantly downloadable and fully editable.
- ✓Complete bundles or individual packages
- ✓45 process packages from €69 each
- ✓ISO 13485 · MDSAP · Combined Kit
Conclusions
The ISO 13485 CAPA procedure is not a compliance checkbox — it is the engine of quality improvement in a medical device organization. When it functions correctly, it identifies systemic problems early, addresses their root causes permanently, and prevents the recurrence of quality events that could harm patients or damage the organization’s regulatory standing. When it functions poorly — closing CAPAs on time without verifying effectiveness, identifying symptoms rather than causes, treating preventive action as an afterthought — it creates the illusion of quality management without the substance.
The organizations that consistently achieve clean audit outcomes on CAPA are not those with the most elaborate software or procedures. They are the ones that invest real time and cross-functional expertise in root cause analysis, that define meaningful effectiveness criteria before implementing actions, and that use CAPA data actively to inform management review and quality strategy.
If you are building or upgrading your CAPA system, the right starting point is a well-structured, role-specific documented procedure that your quality team can actually follow. The ISO 13485 CAPA SOP package available on MD Regulatory includes a complete corrective action procedure (Clause 8.5.2), a preventive action procedure (Clause 8.5.3), a CAPA initiation and investigation form, an effectiveness verification record, and a CAPA trend analysis template — all written to current Notified Body expectations and immediately deployable in your QMS.
This article is part of the MD Regulatory ISO 13485 series. Related articles: ISO 13485 Complete Guide · ISO 13485 Internal Audit Checklist · Benefit-Risk Analysis · MDSAP Audits






2 Comments
Comments are closed.