Software as a Medical Device (SaMD): Classification, Requirements and Regulatory Pathway
Table of Contents
Software as a Medical Device (SaMD) is one of the fastest-growing and most regulated categories in the entire medical device industry. From AI-powered diagnostic algorithms to clinical decision support tools, from mobile apps that analyze ECG data to cloud-based platforms that guide treatment decisions — SaMD is reshaping healthcare delivery while simultaneously navigating one of the most complex regulatory landscapes in the world.
The term Software as a Medical Device is defined by the International Medical Device Regulators Forum (IMDRF) as “software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device.” FDA This definition is now the globally accepted baseline used by the FDA, Health Canada, the EU, and most other major regulatory jurisdictions.
If you are developing a health software product — whether a mobile app, a cloud platform, an AI algorithm, or a clinical decision support tool — this guide will help you understand whether your software qualifies as SaMD, how it will be classified in the EU and the US, and what regulatory requirements you must meet to bring it to market legally and safely.
COMPLETE CATALOG
Find the documentation you need — instantly.
Whether you need a complete kit or just one specific SOP, our catalog has it. 45 process packages and 3 complete bundles, all instantly downloadable and fully editable.
- ✓Complete bundles or individual packages
- ✓45 process packages from €69 each
- ✓ISO 13485 · MDSAP · Combined Kit
SaMD vs SiMD vs MDSW — Getting the Definitions Right
Before entering the classification process, it is essential to understand three terms that are frequently confused.
SaMD — Software as a Medical Device is standalone software that performs a medical purpose independently of any hardware device. It runs on general-purpose computing platforms — smartphones, tablets, cloud servers, desktops — and its medical function does not require a specific hardware medical device to operate. Examples include a mobile app that analyzes retinal images for diabetic retinopathy, a cloud-based algorithm that detects arrhythmias from wearable ECG data, or a clinical decision support tool that recommends treatment protocols based on patient data.
SiMD — Software in a Medical Device is software embedded in or integral to a hardware medical device — the software that runs a ventilator, controls an infusion pump, or drives the image reconstruction in an MRI scanner. Software that is used to power hardware or drive a hardware device does not meet the criteria for SaMD — that type of software is what is known as SiMD. SiMD is regulated as part of the hardware device.
MDSW — Medical Device Software is the broader EU regulatory term used in the MDR and IVDR context, and in MDCG 2019-11 Rev.1. Medical device software is software that is intended to be used, alone or in combination, for a purpose as specified in the definition of a medical device in the MDR or IVDR, regardless of whether the software is independent or driving or influencing the use of a device. MDSW therefore includes both SaMD (standalone) and software that drives or influences hardware devices.
Understanding which category your software falls into is the first and most important step — it determines which classification rules apply and which regulatory pathway you must follow.
Is Your Software a Medical Device? — The Qualification Process
Not all health software is a medical device. Before classification, manufacturers must complete a qualificationassessment — determining whether their software falls within the regulatory scope of the MDR, FDA, or other applicable frameworks.
The key question in qualification is the intended purpose. Software qualifies as a medical device when it is intended to be used for one or more of the following purposes: diagnosis, prevention, monitoring, prediction, prognosis, treatment, or alleviation of disease or injury; investigation, replacement, or modification of anatomy or a physiological process; control or support of conception; disinfection or sterilization of medical devices.
Software that does not qualify as a medical device includes general wellness apps (fitness tracking, calorie counting, meditation tools), administrative software (scheduling, billing, EHR storage with no analytical function), and software that only stores, archives, or communicates data without performing any analytical or decision-support function. An electronic health record system that only stores and retrieves patient records or a patient appointment scheduling system would not be considered MDSW.
The boundary cases — particularly AI-powered wellness apps that approach clinical functionality, and clinical decision support tools — are the most complex to qualify and are the subject of ongoing regulatory guidance updates in both the EU and the US.
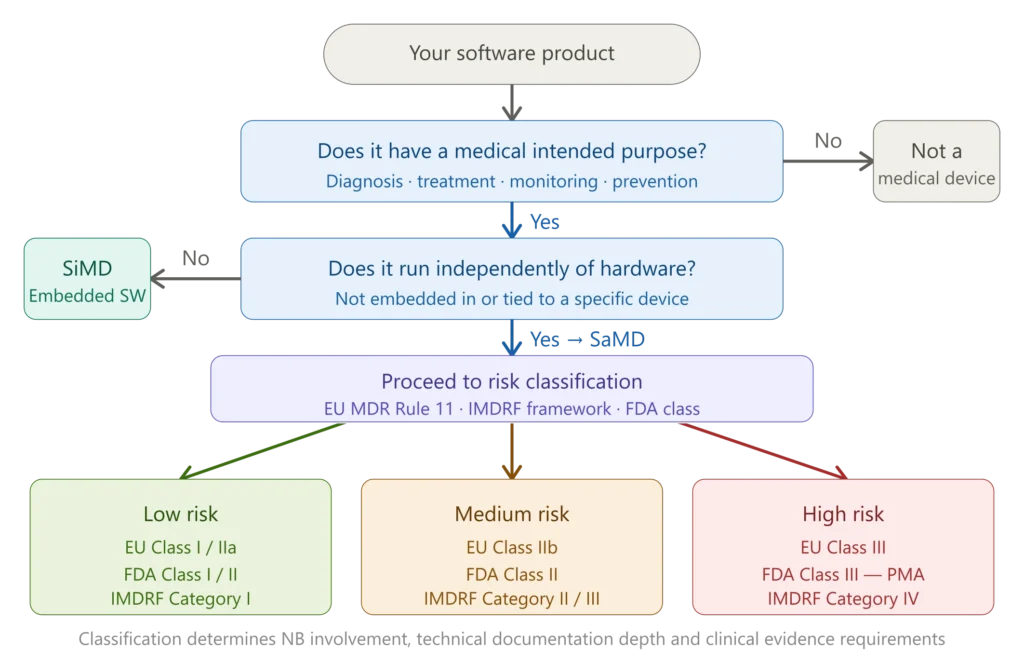
Figure 1 — SaMD qualification and classification flowchart
The IMDRF SaMD Classification Framework
The IMDRF developed the globally recognized risk categorization framework for SaMD in 2014. The IMDRF framework evaluates SaMD across two critical dimensions. Healthcare Decision Information ranges from non-serious (low impact on patient outcomes) to critical (life-threatening condition decisions). Healthcare Situation considers whether use occurs in professional healthcare environments or patient settings outside clinical supervision. This creates four risk categories from Category I (lowest) to Category IV (highest), directly determining documentation requirements and approval pathways.
The two dimensions of the IMDRF framework are:
Significance of information provided by the SaMD:
- Treat or diagnose — the SaMD drives the clinical management decision directly
- Drive clinical management — the SaMD informs but a clinician makes the final decision
- Inform clinical management — the SaMD provides supplementary information
State of the healthcare situation or condition:
- Critical — a life-threatening condition or serious deterioration of health is involved
- Serious — conditions requiring major therapeutic interventions
- Non-serious — conditions not requiring major interventions
The combination of these two dimensions produces four risk categories. Category IV (treat/diagnose + critical situation) represents the highest risk — software that directly drives treatment decisions for life-threatening conditions. Category I (inform + non-serious) represents the lowest risk. The IMDRF framework is not legally binding in itself, but has been adopted by both the FDA and the EU (through MDCG 2019-11 Rev.1) as the conceptual basis for their respective classification systems.
SaMD Classification Under EU MDR — Rule 11 and MDCG 2019-11 Rev.1
In the EU, SaMD is classified as Medical Device Software (MDSW) and falls primarily under Rule 11 of Annex VIII of EU MDR 2017/745. Rule 11 of Chapter III in Annex VIII of the MDR provides that software intended to provide information which is used to take decisions with diagnosis or therapeutic purposes is classified as Class IIa, except if such decisions have an impact that may cause death or an irreversible deterioration of a person’s state of health, in which case it is Class III, or serious deterioration of a person’s state of health or a surgical intervention, in which case it is classified as Class IIb.
A critical practical implication of Rule 11 is that Class I does not appear in the table, suggesting that MDSW under EU MDR Class I will be rare, and software will tend to have a higher risk classification. This means that nearly all SaMD that provides diagnostic or therapeutic information will be classified as at least Class IIa — requiring Notified Body involvement.
MDCG 2019-11 Rev.1 — the June 2025 update introduced important clarifications that every SaMD manufacturer must be aware of. The term Medical Device Artificial Intelligence (MDAI) appears for the first time, reflecting a growing regulatory focus on AI-enabled software as a distinct subset of MDSW. The concept of modular MDSW is reinforced, highlighting the need for each module’s intended purpose to be clearly defined and documented.
Additional examples are added to aid the interpretation of whether MDSW not intended for medical purposes (e.g., wellness and lifestyle apps) still falls under MDR via Annex XVI. An additional example illustrates when MDSW may be justifiably classified as low risk — underscoring that low-risk tools based on validated algorithms with no direct clinical action may confidently be classified as Class I.
The EU classification outcome by device class:
Class I SaMD: Very rare. Only software with negligible risk and no diagnostic/therapeutic decision function. Self-certification, no Notified Body required.
Class IIa SaMD: Most diagnostic and therapeutic decision-support tools where incorrect output leads to non-serious harm. Notified Body QMS review required.
Class IIb SaMD: Tools whose incorrect output may lead to serious harm or require surgical intervention. Full Notified Body assessment including technical documentation review.
Class III SaMD: Life-critical software — tools whose malfunction could cause death or irreversible deterioration of health. Most rigorous review including potential expert panel consultation.
SaMD Classification Under FDA
The FDA classifies SaMD within its standard three-tier device classification system — Class I, II, or III — based on the risk level and the regulatory controls needed to provide reasonable assurance of safety and effectiveness.
FDA regulatory classifications more closely align with the IMDRF’s SaMD categories than those defined under EU MDR. In the US, a device is classified by identifying similar predicate devices. The simpler 510(k) pre-market submission can be used if a manufacturer can show that their device is substantially equivalent to a Class I or II device. Devices where no substantial equivalent can be shown are subject to the PMA or De Novo regulatory submission processes.
FDA regulatory pathways for SaMD:
510(k) — Premarket Notification: The most common pathway for Class II SaMD. Requires demonstration of substantial equivalence to a legally marketed predicate device. FDA review typically takes 3-6 months. The majority of diagnostic and clinical decision support software falls in this category.
De Novo: For novel low-to-moderate risk SaMD with no predicate device. Allows the FDA to create a new device classification. Once granted, the De Novo-cleared device itself becomes a predicate for future 510(k) submissions by others.
PMA — Premarket Approval: Required for Class III SaMD — software that supports or sustains life, or whose failure could cause serious adverse health consequences. Requires valid scientific evidence — including clinical data — to demonstrate safety and effectiveness. Review takes 10-12 months minimum.
An important 2026 development: in January 2026, the FDA updated its guidance on Clinical Decision Support (CDS) software and general wellness products, taking a more deregulatory posture toward certain lower-risk software functions — for example, broadening the scope of non-invasive wearables that qualify as general wellness products exempt from regulation. This means some software that previously fell within FDA’s SaMD scope may now be exempt — a gap analysis against the updated CDS guidance is advisable for manufacturers of clinical decision support tools.

Figure 2 — EU MDR vs FDA SaMD classification comparison
Key Regulatory Requirements for SaMD Manufacturers
Once classification is determined, SaMD manufacturers must meet a comprehensive set of regulatory requirements spanning quality management, software development, risk management, clinical evaluation, cybersecurity, and post-market surveillance.
ISO 13485 — Quality Management System
All SaMD manufacturers placing devices on the EU market or the US market under QMSR must operate a quality management system compliant with ISO 13485:2016. The QMS must cover the entire software development lifecycle — from design inputs through post-market surveillance — with documented processes, records, and controls proportionate to device risk class.
IEC 62304 — Software Lifecycle Processes
IEC 62304 is the primary technical standard for medical device software development. It defines requirements for software development planning, requirements analysis, architectural design, detailed design, implementation, integration and integration testing, system testing, software release, and software maintenance — organized by software safety class (A, B, or C) based on the severity of potential harm if the software fails.
For SaMD, full IEC 62304 compliance is expected by both the FDA (as a recognized consensus standard) and EU Notified Bodies. The IEC 62304 documentation package — including software development plan, software requirements specification, software architecture document, SOUP list, test plans and reports — forms a critical part of the technical documentation for any SaMD above Class I.
ISO 14971 — Risk Management
Risk management under ISO 14971:2019 must be applied throughout the SaMD lifecycle. For software, risk analysis must specifically address software-specific hazards — incorrect output, software failure, cybersecurity vulnerabilities, use errors, and interaction failures. The risk management file feeds directly into the benefit-risk analysis required by EU MDR and FDA.
Clinical Evaluation and Clinical Performance
SaMD requires clinical evidence demonstrating that its output is clinically meaningful, analytically valid, and delivers the claimed clinical benefit. The clinical evaluation of SaMD includes three main pillars: valid clinical association — demonstrating that the SaMD output is clinically relevant and supported by medical evidence; analytical validation — evaluating the technical performance and data processing accuracy; and clinical validation — confirming the SaMD performs as intended in real medical use.
Cybersecurity
Cybersecurity is no longer optional for SaMD — it is a mandatory regulatory requirement in both the EU and the US. EU MDR Annex I GSPR 17 requires that SaMD incorporating programmable electronic systems includes measures to protect against unauthorized access. The primary standard is IEC 81001-5-1. In the US, the FDA’s cybersecurity guidance requires a Software Bill of Materials (SBOM), vulnerability management processes, and a coordinated vulnerability disclosure policy as part of premarket submissions.
SaMD and Artificial Intelligence — The Emerging Frontier
AI-powered SaMD represents the most rapidly evolving area of medical device software regulation. In December 2024, the FDA finalized its guidance on Predetermined Change Control Plans (PCCPs) for AI-enabled device software functions, allowing manufacturers to pre-specify anticipated algorithm updates in their original marketing submission and implement those changes without filing a new application, provided they follow the approved protocol.
Additionally, the FDA issued draft guidance in January 2025 on AI-enabled device software functions applying a Total Product Life Cycle (TPLC) approach.
In the EU, the term Medical Device Artificial Intelligence (MDAI) appears for the first time in MDCG 2019-11 Rev.1, reflecting a growing regulatory focus on AI-enabled software as a distinct subset of MDSW subject to the AI Act.
Manufacturers developing AI-powered SaMD must now navigate a dual regulatory framework — EU MDR for the medical device classification and conformity assessment, and the EU AI Act for AI system obligations — with the intersection of the two becoming mandatory from 2027 onward.
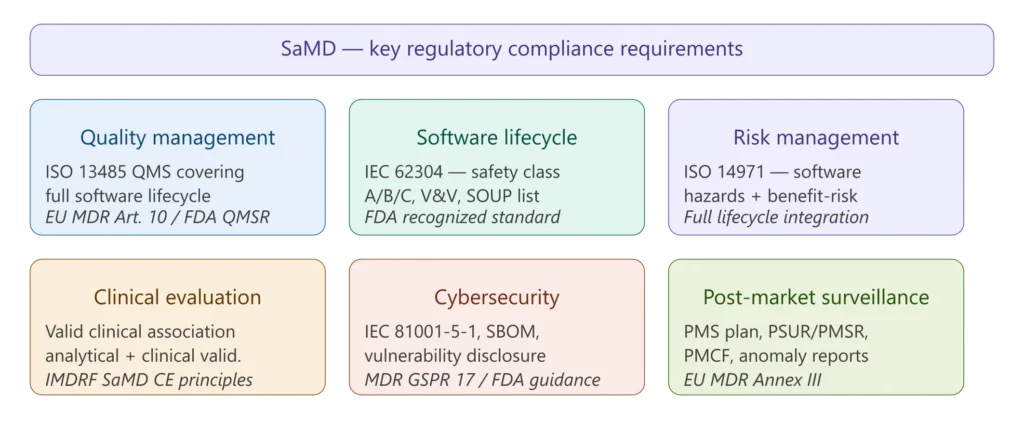
Figure 3 — SaMD key regulatory requirements overview
SaMD Technical Documentation Requirements
Whether you are pursuing CE marking under EU MDR or FDA clearance/approval, SaMD requires comprehensive technical documentation. The depth and scope scale with risk class, but the core structure is consistent.
For EU MDR conformity assessment, the technical documentation must follow Annex II and III of EU MDR and must include a precise intended purpose statement specifying the medical condition, patient population, clinical setting, and the information the software provides; a complete software description including architecture, interfaces, SOUP components, and security measures; IEC 62304 software lifecycle documentation; ISO 14971 risk management file with software-specific hazard analysis; clinical evaluation report demonstrating valid clinical association, analytical validation, and clinical validation; IEC 62366 usability engineering file; IEC 81001-5-1 cybersecurity documentation; and post-market surveillance plan.
For FDA premarket submissions, the content of premarket submissions for device software functions is governed by FDA’s guidance document of the same name. For 510(k) submissions, this typically includes a device description, software level of concern, software description, device hazard analysis, software requirements specification summary, architecture design chart, software design specification summary, traceability analysis, software development and maintenance practices, verification and validation documentation, and revision history.
✦ PREMIUM BUNDLE
The ultimate global QMS documentation bundle.
Combine ISO 13485 + all 5 MDSAP markets in one premium package. Deduplicated structure means you customize each document once — not twice. Save €199 vs buying the kits separately.
- ✓41 SOPs covering both ISO 13485 and MDSAP
- ✓70+ templates with deduplicated structure
- ✓Save €199 vs buying separately
SaMD Post-Market Surveillance
Post-market surveillance for SaMD is substantially more complex than for hardware devices, because software can be updated continuously, new cybersecurity vulnerabilities are discovered constantly, and real-world performance data may differ significantly from pre-market validation results.
Under EU MDR, SaMD post-market surveillance must include systematic collection of complaint data, vigilance reports, literature monitoring, registry data where applicable, and — for higher-risk devices — an active Post-Market Clinical Follow-Up (PMCF) plan. The PSUR must be updated annually for Class III and implantable Class IIb SaMD, and every two years for Class IIa and non-implantable Class IIb.
Software updates require particular attention in the post-market phase. Not all updates require a new regulatory submission — minor bug fixes, security patches, and usability improvements typically do not. However, changes that affect the intended purpose, the clinical algorithm, the user interface in a clinically significant way, or the security posture of the device may constitute a “significant change” requiring regulatory review before release.
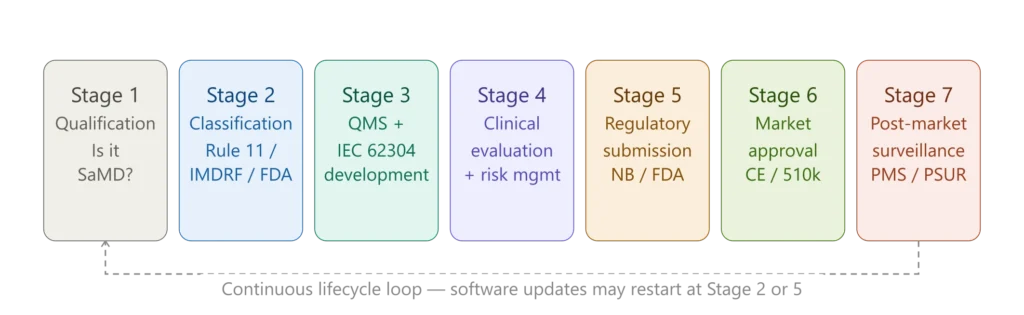
Figure 3 — SaMD key regulatory requirements overview
Practical SaMD Compliance Checklist
For SaMD manufacturers in the early stages of regulatory planning, the following checklist covers the foundational actions required before approaching a Notified Body or submitting to the FDA.
Qualification and classification:
- Confirm your software has a medical intended purpose — document the intended purpose with precision
- Determine whether it is SaMD (standalone) or SiMD (embedded)
- Apply MDCG 2019-11 Rev.1 (EU) or FDA’s software guidance (US) to determine classification
- Identify all applicable classification rules — apply the most stringent where multiple rules apply
QMS and development:
- Establish an ISO 13485-compliant QMS covering all software development activities
- Assign IEC 62304 safety class (A, B, or C) to each software item
- Implement a software development lifecycle per IEC 62304 with full documentation
- Compile and maintain the SOUP list with version control and anomaly tracking
Risk management and clinical evidence:
- Conduct software hazard analysis per ISO 14971, including cybersecurity hazards
- Develop and execute the clinical evaluation plan covering all three IMDRF pillars
- For Class IIb and III EU devices, plan for PMCF from the outset — do not treat it as an afterthought
Cybersecurity:
- Integrate security requirements into the software requirements specification
- Generate an SBOM in a recognized format (CycloneDX or SPDX)
- Establish a coordinated vulnerability disclosure policy and post-market monitoring process
Technical documentation:
- Structure technical documentation to Annex II/III (EU) or FDA submission content guidance (US)
- Ensure traceability between software requirements, design, testing, and risk controls
- Verify cross-document consistency — intended purpose, risk management file, clinical evaluation, and labelling must all tell the same story
Frequently Asked Questions
Is all health software regulated as SaMD? No. Software only qualifies as SaMD if it has a medical intended purpose — diagnosing, treating, monitoring, or preventing a disease or condition. General wellness apps (fitness trackers, calorie counters, stress management tools) and purely administrative software (scheduling, billing, EHR storage without analytical functions) are not SaMD. The boundary cases — particularly AI-powered wellness apps and clinical decision support tools — require careful qualification analysis against the applicable regulatory framework.
Does EU MDR use the term “SaMD”? The term SaMD is not in the EU MDR text, but the concept is fully recognized — any software with a medical intended purpose is regulated under MDR or IVDR requirements just like other devices. The EU uses the term Medical Device Software (MDSW) as defined in MDCG 2019-11 Rev.1.
What is the minimum classification for SaMD under EU MDR? In practice, most SaMD that provides diagnostic or therapeutic information will be classified as at least Class IIa under Rule 11. Class I SaMD is theoretically possible but very rare — it requires that the software provides information that does not influence any diagnostic or therapeutic decision.
Can a mobile app be a medical device? Yes — if it has a medical intended purpose. A mobile app that analyzes ECG data to detect atrial fibrillation is SaMD. A mobile app that tracks daily step count for general wellness is not. The intended purpose — not the platform or form factor — determines regulatory status.
What happens when AI software learns and changes after market release? This is one of the most actively evolving areas of SaMD regulation. The FDA’s Predetermined Change Control Plan (PCCP) guidance, finalized in December 2024, provides a pathway for pre-specifying anticipated algorithm changes so they can be implemented post-market without a new submission. In the EU, any change that affects the intended purpose, safety, or clinical performance of the device requires reassessment before implementation.
Conclusions
Software as a Medical Device sits at the intersection of healthcare innovation and rigorous regulatory oversight — a combination that demands both technical excellence and regulatory literacy from development teams. The IMDRF framework provides a globally harmonized conceptual foundation, but the specific requirements of EU MDR Rule 11 and FDA’s classification system create different practical implications for manufacturers operating in each market.
The single most important investment a SaMD manufacturer can make early in development is a precise, well-documented intended purpose statement — because everything else flows from it: classification, clinical evidence requirements, risk management scope, technical documentation depth, and post-market obligations. Manufacturers who define their intended purpose loosely at the start almost always face reclassification, documentation gaps, and Notified Body findings later.
In January 2026, the FDA updated its guidance on Clinical Decision Support software and general wellness products, taking a more deregulatory posture toward certain lower-risk software functions. Meanwhile in the EU, the intersection of MDR and the AI Act is creating a new compliance landscape. The regulatory environment for SaMD will continue to evolve rapidly — staying current with guidance updates from both the FDA and MDCG is not optional for manufacturers in this space.
This article is part of the MD Regulatory series on medical device software. Related articles: IEC 62304 Complete Guide, SOUP Management under IEC 62304, Medical Device Cybersecurity Testing, AI Medical Device Regulation.