EU MDR Clinical Evaluation Report: Requirements, Structure and Common Mistakes
Table of Contents
The Clinical Evaluation Report is the single most consequential document in a medical device technical file under EU MDR 2017/745. It is also the document most frequently found inadequate by Notified Bodies during conformity assessment — and the one that causes the most delays, the highest remediation costs, and the greatest risk to CE marking timelines.
This guide covers everything manufacturers need to know about EU MDR clinical evaluation report requirements: the regulatory framework, the required structure, the clinical evidence standards, the relationship to MEDDEV 2.7/1 Rev 4, and the most common CER mistakes that result in Notified Body objections.
✦ EU MDR CLINICAL DOCUMENTATION KIT
Clinical evaluation, PMS, PMCF and SSCP — covered end-to-end.
8 Word templates for the full clinical lifecycle under EU MDR — CEP, CER, CDP, PMS Plan, PSUR, PMCF Plan and Evaluation Report, plus SSCP for Class III and implantables. Aligned with MDCG 2020-5/6/7/8/13 and 2022-21.
- ✓8 Word templates · pre-market + post-market
- ✓MDCG-aligned · ready for Notified Body submission
The Regulatory Framework: MDR Article 61 and Annex XIV
The clinical evaluation obligation under EU MDR is established primarily in Article 61 and Annex XIV of Regulation 2017/745. Together they define what clinical evaluation is, what it must demonstrate, and how it must be conducted and documented.
Article 61 establishes the foundational principle: manufacturers must conduct a clinical evaluation to confirm conformity with the relevant general safety and performance requirements set out in Annex I. Clinical evaluation is not optional, not a one-time exercise, and not limited to high-risk devices. It applies to all medical devices, regardless of class.
The key requirements established by Article 61 include:
- Clinical evaluation must be based on clinical data — defined in Annex XIV Part A as data concerning safety or performance generated from the use of the device
- Clinical evaluation must be planned, conducted, and documented according to a defined Clinical Evaluation Plan
- The results must be documented in a Clinical Evaluation Report
- The CER must be updated throughout the lifetime of the device as part of the post-market surveillance system
- For Class III and implantable devices, clinical investigations are generally required unless reliance on existing clinical data can be duly justified
Annex XIV provides the detailed requirements, dividing the clinical evaluation into two parts: Part A covering the clinical evaluation process itself, and Part B covering Post-Market Clinical Follow-up (PMCF).
MEDDEV 2.7/1 Revision 4: The Primary Guidance Document
Despite being published in 2016 — before EU MDR came into force — MEDDEV 2.7/1 Revision 4 remains the primary guidance document for clinical evaluation methodology under EU MDR. Notified Bodies continue to use it as the benchmark for assessing CER quality, and the European Commission has not replaced it with MDR-specific guidance.
MEDDEV 2.7/1 Rev 4 establishes the staged clinical evaluation process (Stages 0–4), the requirements for literature search methodology including systematic literature review principles, the concept of the state of the art and how it must be defined, the standards for clinical data appraisal and weighting, the requirements for CER structure and content, and the concept of clinical equivalence and the conditions under which it can be claimed.
Manufacturers should be aware that while MEDDEV 2.7/1 Rev 4 remains applicable, EU MDR has tightened several requirements beyond what the guidance envisaged — particularly around equivalence, the definition of sufficient clinical evidence, and PMCF obligations. The guidance document is available for download directly from the European Commission website and should be treated as a mandatory reference document for anyone involved in clinical evaluation.
The Clinical Evaluation Process: Four Stages
MEDDEV 2.7/1 Rev 4 structures the clinical evaluation as a four-stage process. Each stage must be documented in the CER.
Stage 0 — Scope Definition
Before collecting any data, the scope of the clinical evaluation must be defined. This includes the device description (intended purpose, intended users, intended patient population, indications, contraindications, and clinical claims), the intended clinical benefits and clinical outcomes used to demonstrate them, the relevant general safety and performance requirements from MDR Annex I that require clinical evidence, and the Clinical Evaluation Plan (CEP) which documents all of the above.
The CEP is a mandatory document under MDR Annex XIV. It must be part of the technical documentation and must be updated whenever the device or its intended purpose changes. A CER without a documented CEP — or with a CEP that was clearly written after the evaluation was conducted — is immediately non-compliant.
Stage 1 — Identification of Pertinent Data
All clinical data relevant to the device and its intended purpose must be identified. Under MEDDEV 2.7/1 Rev 4, clinical data comes from three primary sources: clinical investigations (data generated from formal clinical studies conducted in accordance with MDR Annex XV, providing the highest quality evidence), literature (published scientific data concerning the device, equivalent devices, or the clinical condition it addresses, requiring systematic review methodology), and post-market data (complaint data, vigilance reports, PMS reports, PMCF study results, registry data, and any other real-world evidence from marketed devices).
Stage 2 — Appraisal of Pertinent Data
Each piece of clinical data must be appraised for relevance, quality, and weight. Appraisal requires assessment of methodological quality (study design, sample size, follow-up duration, statistical methods), assessment of relevance to the device under evaluation, assessment of potential bias and confounding factors, and an explicit weighting of each data source in terms of its contribution to the overall evidence. The appraisal must be documented in sufficient detail to allow an independent Notified Body reviewer to follow the reasoning.
Stage 3 — Analysis of the Clinical Data
The appraised clinical data must be analyzed to determine whether it demonstrates that the device achieves the intended clinical benefits, that risks are acceptable when weighed against those benefits, and that the device performs as claimed. This analysis must address clinical performance, clinical safety, benefit-risk, and residual risks requiring further PMCF data collection.
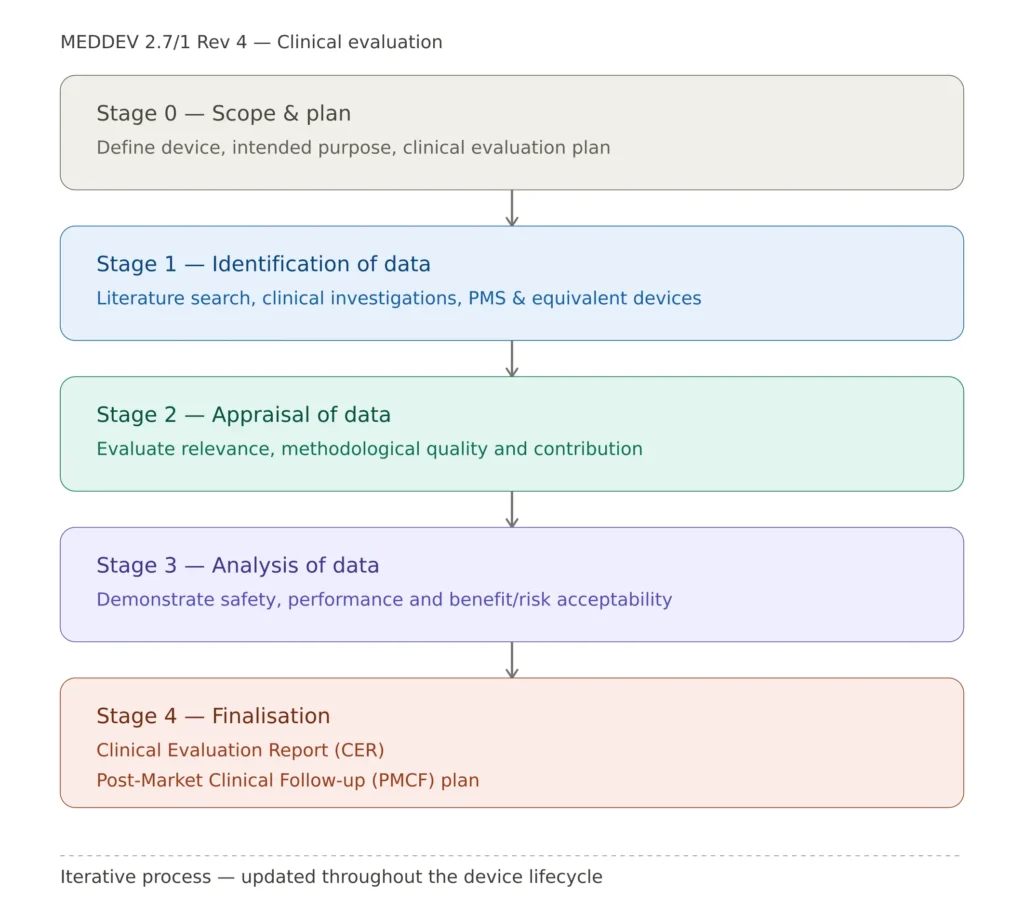
Clinical evaluation process: four stages per MEDDEV 2.7/1 Rev 4 leading to CER and PMCF plan
CER Structure: What Must Be Included
While EU MDR and MEDDEV 2.7/1 Rev 4 do not prescribe a mandatory CER template, the following structure reflects current Notified Body expectations and is the basis on which conformity assessment reviewers evaluate CER quality.
1. Device description and intended purpose — complete device identification including name, model numbers, device generation, intended purpose, indications, contraindications, intended users, intended patient population, and all clinical claims. This section must be consistent with all other technical documentation including the labeling and IFU.
2. Clinical background and state of the art — documented assessment of the clinical condition, current standard of care, alternative technologies, and the state of the art for devices of this type. Must be defined from the perspective of the clinical community, not the manufacturer.
3. Clinical Evaluation Plan — either reproduced in full or incorporated by reference. Must confirm the CEP was in place before the evaluation commenced and was signed and dated prior to Stage 1.
4. Clinical data — device-specific literature — systematic literature review results including search strategy, database list, search terms, date range, inclusion/exclusion criteria, PRISMA flow diagram, and data extraction and appraisal for each included study.
5. Clinical data — equivalent device literature (where applicable) — full documentation of the equivalence claim across technical, biological, and clinical criteria, and confirmation of access to the equivalent device’s technical documentation per MDR Annex XIV requirements.
6. Clinical data — post-market data — systematic analysis of complaint data, vigilance data, PMCF results, registry data, and any other real-world evidence. This section must treat post-market data as a clinical data source, not as an administrative reference.
7. Clinical data appraisal summary — consolidated table of all included clinical data with appraisal ratings and contribution to the overall evidence base.
8. Analysis and conclusions — addressing clinical performance, clinical safety, benefit-risk analysis, and conformity with the applicable GSPRs from MDR Annex I.
9. Residual risks and PMCF rationale — identification of unanswered clinical questions and the rationale for the PMCF plan, including why PMCF activities are sufficient to address those uncertainties.
10. Date, version, and evaluator qualifications — signed and dated, with appended evaluator CV demonstrating relevant clinical expertise.
Clinical Equivalence Under EU MDR: A Critical Change
One of the most significant changes introduced by EU MDR compared to MDD is the tightened requirements for claiming clinical equivalence. Under MDD, basing a CER almost entirely on literature from equivalent or similar devices was common practice. Under EU MDR, this approach is no longer acceptable for most device classes.
MDR Annex XIV Part A Section 3 requires that all three of the following criteria are met simultaneously:
Technical equivalence — same design, same materials in contact with the human body, same principles of operation, same deployment method, same specifications and properties.
Biological equivalence — same materials, same contact with the same tissues or body fluids, same duration and frequency of contact.
Clinical equivalence — same intended purpose, same body site, same clinical condition, same patient population, same user.
For Class III and implantable devices, MDR adds a contractual requirement: the manufacturer claiming equivalence must have a formal contract with the manufacturer of the equivalent device giving access to its technical documentation. This has effectively eliminated equivalence-based CERs for Class III implantables in most cases, as competitors are unwilling to grant such access.
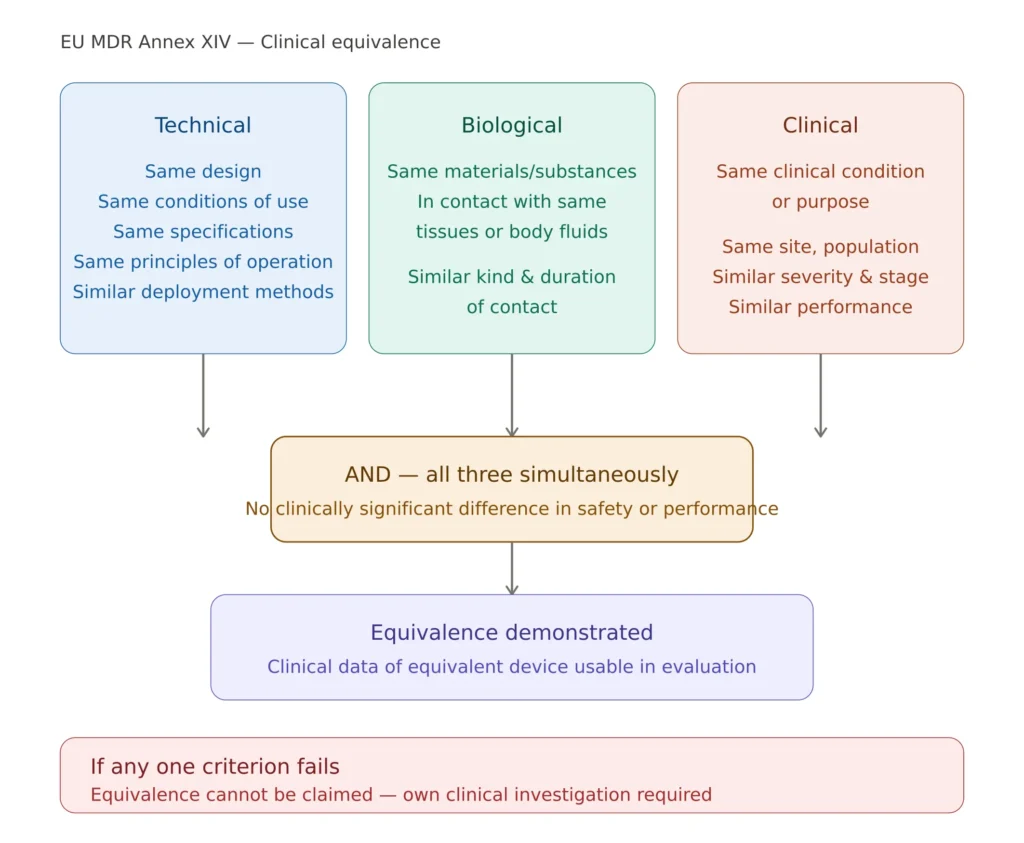
EU MDR clinical equivalence: all three criteria must be demonstrated simultaneously per MDR Annex XIV
PMCF and the CER: An Inseparable Relationship
The Clinical Evaluation Report and the Post-Market Clinical Follow-up plan are inseparable under EU MDR. The CER must identify residual clinical uncertainties that justify PMCF activities, and the PMCF plan must explain how those activities will generate the data needed to update the CER.
This creates a defined lifecycle for clinical evidence. The CER identifies gaps and uncertainties in the available clinical evidence. The PMCF plan defines studies, registries, or surveys designed to address those gaps. PMCF data is collected and analyzed in the PMCF Evaluation Report (PMCFER). The PMCFER feeds back into the updated CER at the next periodic review. The updated CER may identify new uncertainties or confirm that previous ones have been adequately addressed.
For most devices, Notified Bodies expect the CER to be updated at least annually, with the update cycle aligned to the Periodic Safety Update Report (PSUR) for Class IIa, IIb, and III devices. A CER that concludes there are no residual clinical uncertainties and therefore no PMCF is required will face significant Notified Body scrutiny unless the device has an exceptionally long and well-documented market history.
✦ PREMIUM BUNDLE · EU MDR COMPLETE
The complete EU MDR documentation set — technical, clinical and risk.
The Technical Documentation Kit (Annex II), the Clinical Documentation Kit (Articles 32, 61, 83-86) and the Risk Management Kit (ISO 14971 + IEC 62366-1) in one premium bundle. 22 templates, cross-referenced, with a single Master Index.
- ✓22 templates · pre-market + post-market documentation
- ✓Save €178 vs buying the three kits separately
Evaluator Qualifications: A Frequently Overlooked Requirement
MDR Article 61(6) requires that clinical evaluations be carried out by suitably qualified individuals with expertise in the relevant medical field. The qualifications of the evaluator(s) must be documented in the CER — typically through a CV appended to the report. Notified Bodies routinely review evaluator CVs and will raise objections if the evaluator lacks demonstrable expertise in the relevant clinical field.
This has significant practical implications: a regulatory affairs professional without clinical background cannot sign a CER as sole evaluator; a clinical expert without knowledge of MDR requirements needs regulatory support; and many manufacturers engage specialist clinical evaluation consultants or contract research organizations to author or co-author CERs. Evaluator qualification is one of the simplest issues to address in advance — and one of the most avoidable causes of Notified Body objections.
Most Common CER Mistakes That Lead to Notified Body Objections

Most common EU MDR CER deficiencies identified by Notified Bodies during conformity assessment
Based on Notified Body published feedback and industry experience, these are the most frequently cited deficiencies in Clinical Evaluation Reports submitted for EU MDR conformity assessment:
Inadequate literature search methodology — searches limited to a single database, no documented search strategy, no PRISMA flow diagram, no inclusion/exclusion criteria, or cherry-picking of favorable publications. A literature search is not a Google Scholar browse — it must follow systematic review principles with full documentation of every methodological decision.
Insufficient state of the art definition — the state of the art section describes only the manufacturer’s device rather than the broader clinical landscape and alternative technologies. Notified Bodies expect this section to be written from the perspective of the clinical community, benchmarking the device against alternatives.
Unsubstantiated equivalence claims — equivalence claimed without full documentation of all three criteria, or equivalence claimed for Class III devices without the required contract with the equivalent device manufacturer.
Inadequate benefit-risk analysis — the analysis restates claims without quantifying benefits, does not identify specific residual risks, or concludes that benefits outweigh risks without reference to clinical data. The benefit-risk analysis must be evidence-based and device-specific.
Outdated CER — authored at the time of initial CE marking and not updated since, with no incorporation of post-market data or PMCF results. Under EU MDR a static CER is a non-compliant CER.
Missing or disconnected CEP — the Clinical Evaluation Plan is absent, undated, or clearly written after the evaluation was conducted rather than before.
Evaluator qualifications not documented — no CV appended, or CV demonstrates insufficient expertise in the relevant clinical field.
Inadequate appraisal of included studies — papers included without methodological quality assessment, or all papers given equal weight regardless of study design. A case report and a randomized controlled trial cannot contribute equally to a clinical evaluation.
Post-market data not analyzed — complaint data and vigilance information mentioned but not systematically analyzed for clinical trends. Post-market data must be treated as a clinical data source.
Clinical claims not mapped to evidence — claims in the labeling or IFU not supported by specific clinical evidence in the CER. Every clinical claim must be traceable to a clinical data source within the CER.
CER Periodic Update: Frequency and Triggers
Under EU MDR the CER is a living document. It must be updated as part of the annual PSUR cycle for Class IIa, IIb, and III devices; whenever significant post-market safety data becomes available; whenever new literature materially affects the benefit-risk conclusions; whenever the device or its intended purpose is modified; and whenever PMCF results are available from planned studies or registries.
The CER must have a defined review schedule documented in the ISO 13485 Quality Management System, with assigned responsibility and clear criteria for triggering an unplanned update between scheduled reviews. Organizations that fail to maintain a CER update schedule consistently receive findings during Notified Body surveillance audits.
Frequently Asked Questions
Is a CER required for all medical devices under EU MDR? Yes. Article 61 requires clinical evaluation for all devices regardless of class. The depth and rigor varies significantly by device class — a Class I non-sterile device requires a much less extensive CER than a Class III implantable — but no device is exempt from clinical evaluation under EU MDR.
Can a CER be based entirely on literature without clinical investigation data? For Class I and Class IIa devices a literature-based CER may be sufficient if the clinical evidence is adequate to demonstrate conformity with the applicable GSPRs. For Class IIb and Class III devices, Notified Bodies will expect clinical investigation data or a very well-justified rationale for why literature-based evidence is sufficient.
How long does it take to write a compliant CER? For a Class IIb or Class III device, a comprehensive CER typically requires 3–6 months including literature search, data appraisal, clinical analysis, and report writing. Compressing this timeline significantly increases the risk of quality deficiencies that result in Notified Body objections.
What is the difference between a CER and a clinical evaluation? Clinical evaluation is the process — the systematic collection, appraisal, and analysis of clinical data. The CER is the document that records that process and its conclusions. The CER is the output of the clinical evaluation, not the evaluation itself.
Can one CER cover a device family? Yes, in some circumstances where devices share the same intended purpose, clinical mechanism of action, and patient population. Notified Bodies vary in their acceptance of family CERs — this approach should be discussed with the Notified Body before being adopted.
What happens if the Notified Body rejects the CER? The Notified Body issues a list of questions or objections that must be addressed before conformity assessment can proceed. Each remediation iteration adds months and significant cost to the certification timeline — which is why getting the CER right on the first submission is critical.
COMPLETE CATALOG
Find the documentation you need — instantly.
Whether you need a complete kit or just one specific SOP, our catalog has it. 45 process packages and 3 complete bundles, all instantly downloadable and fully editable.
- ✓Complete bundles or individual packages
- ✓45 process packages from €69 each
- ✓ISO 13485 · MDSAP · Combined Kit
Conclusions
The Clinical Evaluation Report is not a bureaucratic exercise — it is the document that demonstrates, with clinical evidence, that a medical device is safe and performs as claimed for its intended population. Under EU MDR the bar for what constitutes a compliant CER has risen substantially compared to MDD, and Notified Bodies are applying that higher standard consistently across all device classes.
The manufacturers who navigate EU MDR conformity assessment most efficiently treat clinical evaluation as a continuous, evidence-based process — starting with a solid Clinical Evaluation Plan, conducting a rigorous systematic literature review, integrating post-market data systematically, and updating the CER on a defined schedule aligned with the PSUR cycle.
For manufacturers building or updating their clinical evaluation documentation, the right starting point is a thorough gap analysis of the existing CER against MDR Annex XIV and MEDDEV 2.7/1 Rev 4 — identifying what clinical evidence is missing, what methodological gaps exist, and what PMCF activities are needed to address residual uncertainties.
The ISO 13485 QMS Documentation Bundle available on MDRegulatory includes post-market surveillance procedures, PMCF templates, and clinical evaluation supporting documentation — giving your quality and regulatory team the foundation needed to maintain a compliant CER update cycle within your QMS.
This article is part of the MD Regulatory series on EU MDR compliance. Related articles cover EU MDR technical documentation requirements, benefit-risk analysis under EU MDR, and ISO 13485 implementation





