Medical Device Cybersecurity Risk Assessment: Threat Modeling and FDA Requirements
Table of Contents
A medical device cybersecurity risk assessment is no longer a documentation exercise that supports a submission — it is now a condition of market access. Since the FDA’s authority under Section 524B of the FD&C Act became effective on 29 March 2023, and with the final premarket cybersecurity guidance issued in June 2025 and terminology aligned to the QMSR in February 2026, a “cyber device” submission that lacks an adequate threat model, security risk assessment or Software Bill of Materials can be refused outright. In the EU, the same expectation arrives through EU MDR Annex I and the MDCG 2019-16 guidance. This guide explains how to perform a medical device cybersecurity risk assessment that satisfies both regimes: scoping and asset identification, data flow diagrams, STRIDE threat modeling, risk estimation, control mapping, and how the whole thing connects to your ISO 14971 safety risk file.
It is written for quality, regulatory and security professionals who already operate a quality management system and need the cybersecurity risk assessment to be submission-proof rather than merely present. It completes the cybersecurity cluster alongside our IEC 81001-5-1 cybersecurity guide and our medical device cybersecurity testing article, and it depends throughout on the safety risk framework described in our ISO 14971 risk management guide.
Why Cybersecurity Risk Assessment Is Now a Gate, Not a Formality
For years cybersecurity in medical devices was treated as advisory — a section auditors might probe but rarely blocked a submission over. That era has ended. The FDA’s June 2025 final guidance, Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions, operationalises Section 524B and makes cybersecurity an independent regulatory standard: a device with unresolved cybersecurity risk can be considered to lack a reasonable assurance of safety and effectiveness on cybersecurity grounds alone. Both the FDA and EU Notified Bodies now reject submissions for cybersecurity shortcomings — a missing SBOM, an inadequate threat model, or absent post-market surveillance planning — independently of any other deficiency.
The definition of which devices are in scope is deliberately broad. A “cyber device” under Section 524B is, in essence, any device that includes software, can connect to the internet (even unintentionally, including dormant wireless modules or hardware ports), and has technological characteristics that could be vulnerable to cybersecurity threats. The practical consequence is that connectivity capability, not active network use, triggers the requirement — a device with a disabled Bluetooth radio or an unused USB port is still a cyber device.
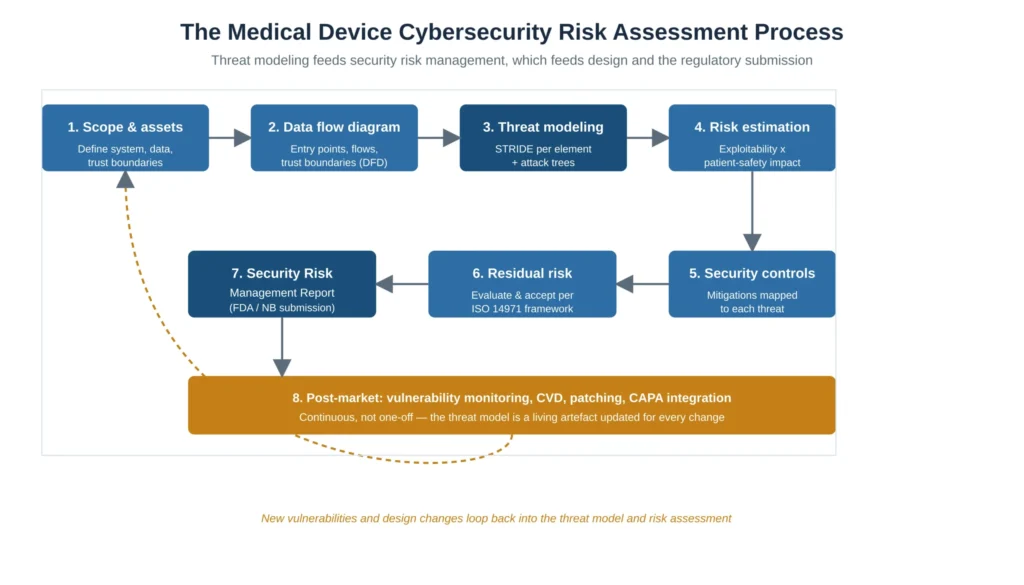
Figure 1 — The medical device cybersecurity risk assessment process: threat modeling feeds security risk management, which feeds design and the regulatory submission
AUDIT-READY KIT
Build your ISO 13485 QMS with confidence.
Built on 15+ years of audit experience — every SOP and template references the regulations auditors expect. Get to certification faster, with industry best practices baked in.
- ✓30 SOPs covering the full QMS scope
- ✓56 templates ready to customize
- ✓Aligned with EU MDR + FDA QMSR
Step 1: Scope, Assets and Trust Boundaries
Every defensible cybersecurity risk assessment begins by defining what is being assessed. This is the step most often done implicitly and most often the root cause of an incomplete threat model later. The scope must identify the device system boundary including any companion app, cloud backend, update server and accessories; the data the device handles, classified by sensitivity (protected health information, device telemetry, configuration, cryptographic material); the interfaces and entry points (wireless radios, physical ports, APIs, update mechanisms, debug interfaces); and the trust boundaries — the lines across which the level of trust changes, such as between the device firmware and a network, or between a clinical user and a service technician.
The FDA’s expectation, and the IEC 81001-5-1 approach, is that this scope explicitly includes “related systems” — the update server and the supporting infrastructure are part of the attack surface, not external to it. A scope that stops at the physical device understates the threat model and is a recurring submission deficiency. The intended clinical environment also belongs here: the same device in a home-use context and a hospital network has different threats, and the assessment must state which environment it assumes.
Step 2: The Data Flow Diagram
The data flow diagram (DFD) is the artefact that makes threat modeling systematic rather than anecdotal. It depicts the processes, data stores, data flows, external entities and — critically — the trust boundaries of the system. STRIDE is applied per element of the DFD, so an incomplete or absent DFD inevitably produces an incomplete threat model. The DFD does not need to be elaborate; it needs to be complete with respect to entry points and trust boundaries, because every place a data flow crosses a trust boundary is where threats concentrate.
A common and serious error is to draw the DFD at a single level of abstraction that hides the interfaces where attacks actually occur — for example, representing “the cloud” as one box without showing the authentication boundary between the device and the cloud API. The DFD should be decomposed until every entry point and every trust boundary that an attacker could target is visible as a distinct element.
Step 3: STRIDE Threat Modeling
STRIDE is the threat modeling method explicitly recognised by both FDA premarket guidance and the EU MDCG 2019-16 / IEC 81001-5-1 framework as an acceptable approach. It is a mnemonic for six categories of threat, each corresponding to a security property that the threat violates. Applying STRIDE means walking every element of the DFD and asking, systematically, whether each of the six threat types applies to it. Figure 2 sets out the six categories with a worked example for a connected infusion pump and the typical control for each.
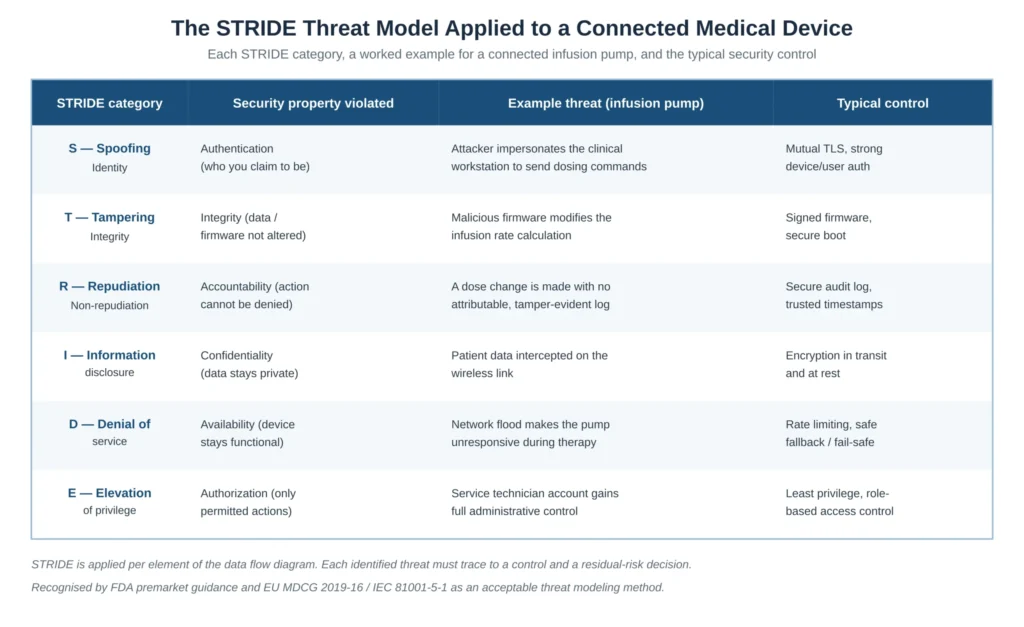
Figure 2 — The STRIDE threat model applied to a connected medical device, with the security property violated, an example threat and the typical control
STRIDE is rarely the whole story. The guidance and good practice expect the threat model to be supplemented where the device characteristics warrant it — attack trees to explore how a high-consequence threat could be reached through chained weaknesses, and consideration of supply-chain and third-party component threats. The output of this step is a threat list in which every threat is attributable to a specific DFD element and a specific STRIDE category, so that completeness can be demonstrated rather than asserted.
Step 4: Risk Estimation — Exploitability Against Patient Safety
This is the step where medical device cybersecurity diverges sharply from general IT security, and where IEC 81001-5-1 deliberately goes beyond a traditional security approach. The risk of a threat is not estimated on business impact or data value; it is estimated on the combination of how exploitable the vulnerability is and what the patient-safety consequence would be if it were exploited. A confidentiality breach of non-sensitive telemetry may be a low risk; a tampering threat that could alter a therapeutic dose is a high risk even if exploitation is difficult, because the harm is severe.
The practical method most aligned with regulator expectations is to assess exploitability using a security scoring approach (for example a CVSS-style analysis adapted to the clinical context) while assessing the impact in terms of patient harm, not data loss. This is the explicit bridge to the safety world: the cybersecurity risk assessment estimates likelihood with security methods but expresses consequence in the same harm terms used by the ISO 14971 risk file.
Step 5: Security Controls and Step 6: Residual Risk
Every threat that is not accepted at its initial risk level must be mapped to one or more security controls — authentication, cryptography, secure boot, least-privilege access, secure logging, network segmentation, and so on, as illustrated in the control column of Figure 2. The control is not credible unless it is both specified as a design input and later verified by security testing; a control claimed in the risk assessment but never tested is one of the most common deficiencies (covered in depth in our medical device cybersecurity testing guide).
After controls are applied, the residual security risk must be evaluated and an explicit accept/reject decision recorded, using the organisation’s risk acceptability criteria. This decision belongs in the same framework as safety risk: residual cybersecurity risk that translates into residual patient-safety risk is governed by the ISO 14971 risk management file, not a separate and disconnected security register.
How Security Risk Connects to the ISO 14971 Safety Risk File
The single most consequential conceptual point in a medical device cybersecurity risk assessment is that it is not a standalone activity. A security threat only matters, in a regulatory sense, to the extent it can lead to a hazardous situation and patient harm. The cybersecurity risk assessment is therefore a parallel process that feeds the safety risk file: the threat model identifies what could go wrong, and the ISO 14971 file governs the safety consequence and its acceptability. Figure 3 shows this linkage and where each piece of evidence ultimately lands in the FDA submission and the EU technical documentation.
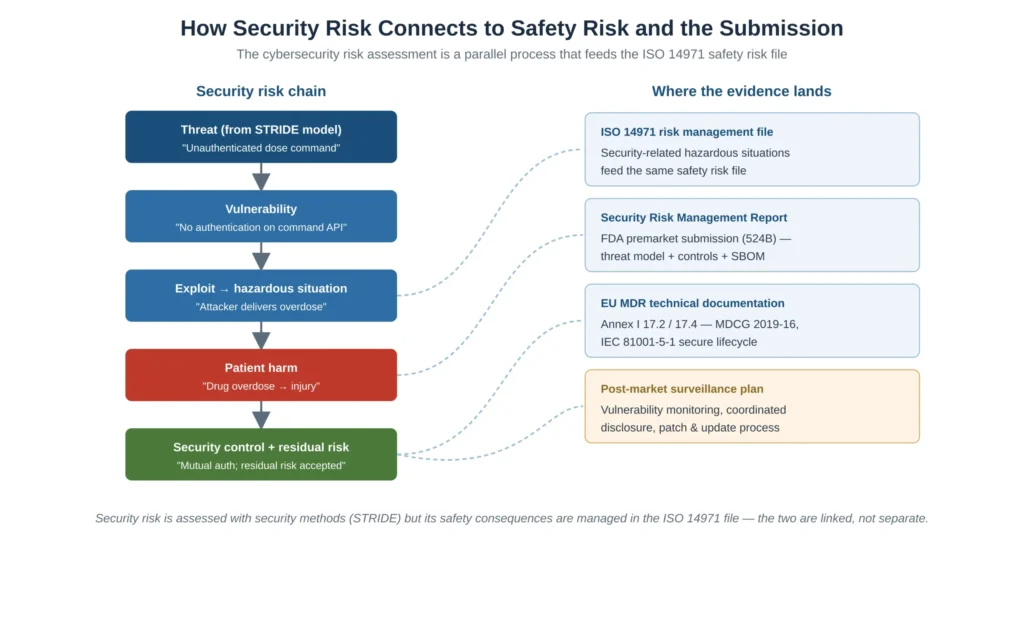
Figure 3 — How security risk connects to safety risk and the regulatory submission; the cybersecurity risk assessment feeds the ISO 14971 safety risk file
FDA and EU Requirements: One Assessment, Two Submissions
The cybersecurity risk assessment is performed once but must satisfy two regulatory regimes that overlap heavily while differing in detail.
On the FDA side, Section 524B of the FD&C Act is the binding statute and the June 2025 final guidance (with February 2026 terminology aligned to the QMSR) is the recommendation framework. The premarket submission for a cyber device must include a security risk management report demonstrating threat modeling and risk analysis, a Secure Product Development Framework (SPDF) spanning the total product lifecycle, security architecture and control documentation, security testing evidence, a machine-readable Software Bill of Materials, and a post-market cybersecurity management plan with coordinated vulnerability disclosure. The FDA can issue a “Refuse to Accept” decision where these are inadequate, and post-market non-compliance is a prohibited act under Section 301(q). The detail of the cybersecurity-specific QMS expectations is developed in our IEC 81001-5-1 cybersecurity guide, and the broader US pathway context is in our FDA De Novo pathway article.
On the EU side, the obligation arrives through EU MDR Regulation (EU) 2017/745 Annex I, particularly sections 17.2 and 17.4, which require development according to the state of the art with IT security considered and protection against unauthorised access. MDCG 2019-16 (Rev. 1) is the current guidance against which Notified Bodies assess, and IEC 81001-5-1 — a secure-lifecycle standard derived from IEC 62443-4-1 and complementing IEC 62304 and IEC 82304-1 — is increasingly the benchmark, with a defined transition period and a transitional assessment route for legacy devices. The cybersecurity evidence becomes part of the broader technical file described in our EU MDR technical documentationguide.
A practical reconciliation point: the threat model, the security risk assessment and the SBOM are common to both submissions. The differences are largely in packaging and in specific expectations around secure communication, update mechanisms and post-market timelines, not in the underlying risk assessment method.
Post-Market: The Assessment Is Never Finished
A cybersecurity risk assessment that is performed once at design and never revisited is non-compliant by construction. Section 524B imposes ongoing obligations to monitor for vulnerabilities and to make updates and patches available across the device lifecycle; the EU framework, through MDCG 2019-16 and post-market surveillance expectations, requires the same. The threat model is therefore a living artefact: it must be re-assessed when the design changes, when a new vulnerability is disclosed in a third-party component listed in the SBOM, and as part of routine vulnerability intelligence. Security events must be integrated into the existing corrective and preventive action process rather than handled in a parallel, disconnected workflow — the discipline described in our ISO 13485 CAPA procedure guide applies directly.
✦ PREMIUM BUNDLE
The ultimate global QMS documentation bundle.
Combine ISO 13485 + all 5 MDSAP markets in one premium package. Deduplicated structure means you customize each document once — not twice. Save €199 vs buying the kits separately.
- ✓41 SOPs covering both ISO 13485 and MDSAP
- ✓70+ templates with deduplicated structure
- ✓Save €199 vs buying separately
Common Cybersecurity Risk Assessment Audit Findings
The same deficiencies recur across FDA submission reviews and EU Notified Body assessments. Figure 4 sets out the six most common, colour-coded by typical severity, each with the corrective approach. Three of them — no structured threat model, security risk divorced from safety risk, and a missing or stale SBOM — are routinely submission-blocking because each prevents the regulator from concluding that cybersecurity risk has been controlled at all.

Figure 4 — Six common cybersecurity risk assessment audit findings, severity-coded, with the fix for each
The pattern across all six is identical to the pattern in design controls generally: the activity may have been done, but the structured, traceable evidence linking threat to control to residual risk to patient harm was never created. A reviewer cannot credit security work that is not demonstrable end to end.
Frequently Asked Questions
What is a cyber device under FDA Section 524B?
A cyber device is, broadly, any device that includes software (including firmware or programmable logic), can connect to the internet, and has technological characteristics that could be vulnerable to cybersecurity threats. The FDA interprets connectivity very broadly: Wi-Fi, cellular, Bluetooth, RF and even hardware connectors such as USB or serial ports count, and the capability rather than active use is what triggers the requirement. A device with a dormant or disabled wireless module is still a cyber device.
Is threat modeling mandatory for medical device submissions?
In practical terms, yes. The FDA’s June 2025 final guidance expects a threat model as part of the security risk management documentation, and a submission without one is a recognised deficiency that can lead to a Refuse to Accept decision. The EU MDCG 2019-16 and IEC 81001-5-1 framework similarly expects a structured threat analysis. STRIDE is explicitly recognised as an acceptable method, though it is not the only one.
What is the difference between security risk and safety risk?
Security risk concerns the likelihood and impact of a threat being exploited; safety risk concerns the probability and severity of harm to the patient. They are linked but assessed differently: security risk uses security methods such as STRIDE and exploitability scoring, while the safety consequence of an exploited threat is managed in the ISO 14971 risk management file. A cybersecurity risk assessment that never connects an exploitable threat to a hazardous situation and patient harm is incomplete.
Is an SBOM required, and what happens if it is missing?
A machine-readable Software Bill of Materials is required in FDA premarket submissions for cyber devices under Section 524B. A missing SBOM, or one that does not correspond to the software actually shipped, is submission-blocking. The SBOM should be generated from the build pipeline and kept synchronised with every release, because a stale SBOM is treated as equivalent to a missing one.
Does the cybersecurity risk assessment ever finish?
No. Section 524B imposes lifecycle obligations to monitor for vulnerabilities and to provide updates and patches, and the EU framework imposes equivalent post-market expectations. The threat model and risk assessment must be re-evaluated when the design changes, when a vulnerability is disclosed in a component listed in the SBOM, and as part of routine vulnerability intelligence, with security events fed into the CAPA process.
Which standards should a cybersecurity risk assessment reference?
The core references are ISO 14971 for the safety risk framework, IEC 81001-5-1 for the secure product lifecycle (complementing IEC 62304 and IEC 82304-1), MDCG 2019-16 for EU expectations, and the FDA June 2025 premarket cybersecurity guidance implementing Section 524B for the US. STRIDE is the most widely recognised threat modeling method; AAMI TIR57 is a useful additional reference for security risk management practice.
Conclusions
A medical device cybersecurity risk assessment is now a gate to market, not a supporting document. The practical takeaways are consistent: scope the system to include related systems and the intended clinical environment; build a data flow diagram complete with trust boundaries; apply STRIDE per element so completeness is demonstrable; estimate risk on exploitability against patient-safety impact, not data value; map every retained threat to a tested control and a recorded residual-risk decision; connect security risk explicitly to the ISO 14971 safety risk file; package the same assessment for both the FDA Section 524B submission and the EU MDR technical documentation; and treat the threat model as a living artefact maintained through post-market vulnerability intelligence and the CAPA process. Get this structure right and cybersecurity stops being the deficiency that blocks your submission and becomes the evidence that most clearly demonstrates the device is safe in a connected world.
The FDA Regulatory Compliance Documentation Package on MD Regulatory includes a security risk management procedure, a threat modeling and STRIDE worksheet template, a security risk assessment and residual-risk record, an SBOM management procedure, and a post-market cybersecurity management plan with coordinated vulnerability disclosure — all aligned with FDA Section 524B and EU MDR / MDCG 2019-16 requirements and immediately deployable in an existing ISO 13485 quality management system.
Related articles: IEC 81001-5-1 Cybersecurity; Medical Device Cybersecurity Testing; ISO 14971 Risk Management; EU MDR Technical Documentation; ISO 13485 CAPA Procedure.

