ISO 14971 Risk Management for Medical Devices: Complete Process Guide
Table of Contents
ISO 14971 is the foundational international standard for risk management of medical devices. It specifies terminology, principles, and a comprehensive process for risk management of medical devices, including software as a medical device and in vitro diagnostic medical devices — establishing a systematic framework for identifying hazards, estimating and evaluating the associated risks, controlling these risks, and monitoring the effectiveness of the controls throughout the entire device lifecycle.
For any medical device manufacturer seeking market access in the EU, USA, Canada, Japan, or Australia, ISO 14971 is not merely best practice — it is the universally expected framework that underpins regulatory compliance across every major market. The FDA, Health Canada, EU Competent Authority, Australia TGA, and Japan MHLW all require you to have a risk management process defined and risk management documentation for your products — and all endorse ISO 14971.
This complete guide covers the ISO 14971:2019 process in full — from planning through hazard identification, risk estimation, risk controls, benefit-risk analysis, and post-market monitoring — with practical guidance on building an audit-ready risk management file. If you are new to the ISO 13485 quality management system that provides the QMS framework within which ISO 14971 operates, we recommend reading that guide first.
What Is ISO 14971 and Why the 2019 Edition Matters
ISO 14971 was first published in 1998 and has been through three editions. The third edition, published in December 2019, superseded the second edition of ISO 14971:2007. Key changes include a stronger emphasis on benefit-risk analysis, more detailed requirements for production and post-production activities, and the introduction of the concept of medical benefit as a defined term.
The 2019 edition is critically important for EU manufacturers because EN ISO 14971:2019 has been officially recognized as a harmonized standard with EU MDR 2017/745 and IVDR 2017/746, eliminating the discrepancies between the global ISO version and the previous EN ISO 14971:2012 European version — which had created compliance complexity by having requirements that differed from the international standard.
The 2019 standard introduces a number of fundamental changes that many manufacturers still have not fully implemented — particularly the benefit-risk analysis requirement, which requires manufacturers to perform a benefit-risk analysis when risks cannot be reduced to acceptable levels through design or protective measures.
ISO/TR 24971:2020 is the companion technical report published in parallel with ISO 14971:2019. It is not normative — it does not contain requirements — but it provides detailed guidance, worked examples, and clarifications for implementing the standard. For any manufacturer building or reviewing their risk management process, ISO/TR 24971:2020 is an essential reference alongside the standard itself.
✦ RISK MANAGEMENT KIT
A defensible risk file under ISO 14971 — with FMEAs that calculate the risk for you.
6 coordinated templates covering the full risk process: Risk Management Plan and Report, Hazard Analysis (with all 46 ISO/TR 24971 Annex A questions), active Excel Design FMEA and Use-related FMEA, and a complete GSPR Checklist mapping all 159 MDR Annex I requirements.
- ✓6 templates · Word + active Excel FMEAs (auto-RPN)
- ✓ISO 14971:2019 + IEC 62366-1 + MDR Annex I aligned
Key ISO 14971 Terminology
Before entering the process, understanding the precise definitions used in ISO 14971 is essential — because many common terms carry specific technical meanings that differ from everyday usage.
Hazard: A potential source of harm. A hazard exists independently of any device — it is a property of the device or its environment. Examples: electrical current, sharp edge, biological agent, software failure, ionizing radiation.
Hazardous situation: The circumstance in which people, property, or the environment are exposed to one or more hazards. A hazardous situation is the link between a hazard and potential harm — it arises from a specific sequence of events.
Harm: Injury or damage to the health of people, or damage to property or the environment. The 2019 edition notably removed the word “physical” from the definition, broadening harm to include psychological injury and data harm.
Risk: The combination of the probability of occurrence of harm and the severity of that harm. Risk has two dimensions — probability and severity — and both must be estimated and combined.
Residual risk: The risk remaining after risk controls have been implemented.
Overall residual risk: The combined residual risk from all identified hazards across the entire device, after all controls have been applied.
Benefit: The positive impact of a medical device on health outcomes — clinical benefit, quality of life, or public health. New to the 2019 edition as a defined term.
ISO 14971 defines safety as “freedom from unacceptable risk.” There is no device that is completely risk-free — the concept of acceptable risk is always relative to the benefit that the device provides. Life-saving devices for otherwise incurable conditions can tolerate a higher risk than devices used for simpler applications.
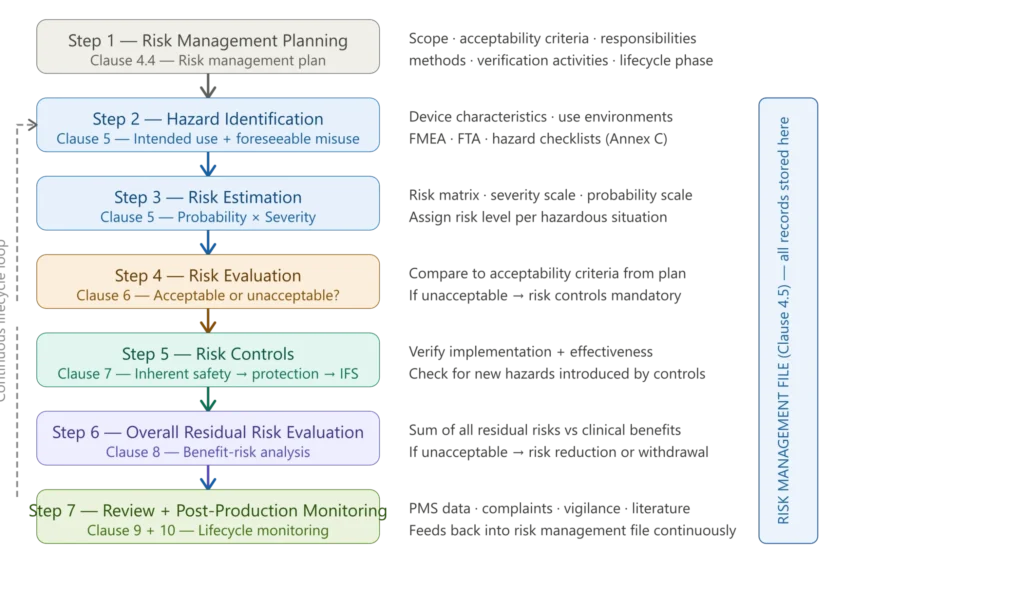
Figure 1 — ISO 14971 risk management process overview
The ISO 14971 Risk Management Process — Step by Step
Step 1 — Risk Management Planning (Clause 4.4)
The starting point of the entire ISO 14971 process is the risk management plan — a document that defines the scope, responsibilities, and methodology for all risk management activities for a specific device or device family. The plan is not a template to be reused across all products; it must be device-specific.
A compliant risk management plan must include: the scope of the plan (which device, which lifecycle phases, which hazard categories are included); the responsibilities and authorities for risk management activities; the criteria for risk acceptability — the manufacturer’s policy for determining when a risk is acceptable, expressed as objective criteria; verification activities for each risk control; the methods to be used for risk analysis; and a plan for production and post-production activities.
The risk management plan is a living document — it must be updated as the design, regulations, or clinical practice changes. Your procedure is not adequate as a risk management plan. You need risk management plans that are product-specific or specific to a product family.
The risk acceptability criteria defined in the plan are among the most scrutinized elements in Notified Body technical documentation reviews. Criteria must be objective and measurable — a statement that “all risks shall be as low as reasonably practicable” without a defined scale is not compliant with the 2019 edition.
Step 2 — Hazard Identification (Clause 5)
Hazard identification is the process of systematically identifying all hazards associated with the device — across all characteristics relevant to safety, all intended use scenarios, and all reasonably foreseeable misuse scenarios.
ISO 14971:2019 requires that hazard identification consider:
- The intended use of the device and reasonably foreseeable misuse
- All characteristics of the device that could affect safety, including physical, chemical, biological, and software characteristics
- The use environment — clinical setting, lay user vs professional user, pediatric vs adult population
- All lifecycle phases — design, manufacturing, transport, storage, installation, use, maintenance, and disposal
Practical tools for hazard identification: Manufacturers can identify hazards by answering questions from a checklist relating to device safety (ISO/TR 24971:2020 Annex A), or by checking the applicability of hazards from a catalogue checklist (ISO 14971:2019 Annex C). Annex C of the standard lists common hazard categories — energy hazards (electrical, mechanical, thermal, radiation), biological and chemical hazards, information hazards (inadequate labelling, incorrect output), and operational hazards.
For medical device software, hazard identification must additionally consider software-specific failure modes — incorrect outputs, failure to perform a required function, timing failures, and cybersecurity vulnerabilities. This connects directly to the software hazard analysis requirements of IEC 62304, which defines how software items of medical devices must be analyzed for safety-related failures.
Hazard vs Hazardous Situation: One of the most common conceptual errors in risk documentation is conflating a hazard with a hazardous situation. A hazard is a potential source of harm that exists intrinsically in the device or environment. A hazardous situation is the specific sequence of events that leads to a patient or user being exposed to that hazard. Both must be documented separately in the risk management file.
Step 3 — Risk Estimation (Clause 5)
For each hazardous situation identified, the manufacturer must estimate the associated risk by assigning values to its two components: the probability of occurrence of harm and the severity of harm.
Severity scale: ISO 14971 does not mandate a specific severity scale — manufacturers define their own in the risk management plan. Common approaches use three to five levels, typically defined qualitatively (negligible, minor, serious, critical, catastrophic) with quantitative anchors or clinical descriptions at each level.
Probability scale: Similarly, manufacturers define their own probability scale. Where historical data exists (from similar devices, clinical literature, or industry databases), probability should be estimated quantitatively. Where data is insufficient, qualitative descriptors are acceptable but must be defined objectively.
Risk matrix: The combination of severity and probability produces an initial risk level, typically represented in a risk matrix. The risk matrix serves as the visual tool for mapping estimated risks to acceptability zones — acceptable, ALARP (as low as reasonably practicable), or unacceptable.
Define scales that engineers and clinicians can actually use. Pre-define what requires escalation — for example, any catastrophic harm with credible occurrence — and what needs management sign-off. Bake those thresholds into your risk register and SOPs so decisions are consistent and auditable.
✦ BUNDLE · RISK + CLINICAL
The clinical and risk file for EU MDR — designed to reference each other.
The Risk Management Kit and the EU MDR Clinical Documentation Kit, bundled together. The Risk Management Report cross-references the CER for benefit-risk conclusions, and the GSPR Checklist references both clinical and risk evidence for each requirement.
- ✓14 templates · risk + clinical, cross-referenced
- ✓Save €149 vs buying the kits separately
Step 4 — Risk Evaluation (Clause 6)
Risk evaluation is the decision step — comparing each estimated risk against the acceptability criteria defined in the risk management plan, and determining whether risk reduction is required.
Three outcomes are possible:
Risk is acceptable: The risk as estimated falls within the acceptable zone defined in the plan. No risk controls are required from a risk management perspective, though the risk must still be documented in the risk management file and included in the overall residual risk evaluation.
Risk requires reduction (ALARP zone): The risk may be tolerable if it cannot be reduced further, but the manufacturer must make every reasonable effort to reduce it. Risk controls must be investigated and implemented unless the reduction is not technically practicable.
Risk is unacceptable: Risk controls are mandatory. The device cannot be placed on the market unless the risk can be reduced to an acceptable level.
An important distinction in the 2019 edition: under EU MDR, the standard requires that all risks be reduced as far as possible — not just risks above the acceptability threshold. Some individuals following EU MDR say that a benefit-risk analysis is required for all risks. The rationale is that the regulation states that all risks shall be reduced as far as possible. GetApp This is a stricter interpretation than the base ISO 14971 standard and should be considered when defining the risk management plan for EU MDR-covered devices.
Step 5 — Risk Controls (Clause 7)
When risk reduction is required, ISO 14971 mandates a hierarchical approach to risk control selection — moving from the most effective to the least effective option:
Priority 1 — Inherent safety by design: Eliminate or reduce hazards through design choices — using safer materials, removing sharp edges, implementing software interlocks, selecting non-toxic coatings. This is always the preferred approach because it removes the hazard at source.
Priority 2 — Protective measures in the device or manufacturing process: Physical guards, safety mechanisms, automatic shutoffs, software error detection, alarms, and interlocks that cannot be bypassed.
Priority 3 — Information for safety (IFS): Warnings, contraindications, precautions, and instructions in labelling and IFU. This is the least effective risk control option and should only be used when inherent safety and protective measures are not practicable or are insufficient.
To provide written information that highlights and clearly discusses a use-related hazard, the manufacturer can provide warning or caution statements in the user manual, train users to avoid the use error, or use specific connectors that cannot be connected to the wrong component.
Verification of risk controls must demonstrate two things: that the risk control was correctly implemented (implementation verification), and that it actually reduces the risk to the intended level (effectiveness verification). Both must be documented with objective evidence — test reports, analyses, usability validation results, or clinical data.
New hazards introduced by risk controls: After implementing any risk control, the manufacturer must check whether the control introduces new hazards or changes the risk estimation of previously identified hazards. If so, those new hazards must be entered into the risk analysis and processed through the full risk management cycle. This creates an iterative character to the process that many manufacturers underestimate in their initial risk management planning.
For devices incorporating software, risk controls may include software-specific measures — algorithm-level safeguards, input validation, range checking, or cybersecurity controls. The cybersecurity risk controls required by IEC 81001-5-1integrate directly into the ISO 14971 risk management framework as a specialized category of risk control for cybersecurity hazards.

Figure 2 — ISO 14971 risk control hierarchy
Step 6 — Benefit-Risk Analysis and Overall Residual Risk Evaluation (Clause 8)
This is one of the most significant additions of ISO 14971:2019 and one of the most frequently misunderstood elements of the standard. It operates at two levels:
Individual benefit-risk analysis: When a risk cannot be reduced to an acceptable level through any practicable risk control measures, but the device provides significant clinical benefit, a benefit-risk analysis may be used to justify accepting the residual risk. The clinical benefits must demonstrably outweigh the residual risk. This is explicitly not a license to avoid risk reduction — all practicable risk controls must still be implemented before benefit-risk analysis is applied to any remaining residual risk.
Overall residual risk evaluation: After all individual risk controls have been applied and verified, the manufacturer must evaluate the combined residual risk from all identified hazards — the overall residual risk of the device. If the overall residual risk is unacceptable even after all controls, the device cannot be placed on the market unless the clinical benefits clearly outweigh the combined residual risks.
For EU MDR-covered devices, this evaluation connects directly to the benefit-risk analysis required under MDR Article 10 and Annex II Section 4. The EU MDR benefit-risk analysis must be supported by clinical evidence — not theoretical estimates — and must be documented in the technical file. Our detailed guide to benefit-risk analysis covers this in depth.
The overall residual risk evaluation must also account for:
- State of the art — the prevailing level of risk acceptability for similar devices on the market and in clinical practice
- Risk-benefit information from published clinical literature and post-market data from similar devices
- Any relevant guidance documents from regulators or professional societies
Step 7 — Risk Management Review and Risk Management Report (Clause 9)
Before placing the device on the market, a formal risk management review must confirm three things:
- The risk management plan has been fully implemented
- The overall residual risk is acceptable
- Procedures are in place to gather, review, and act on production and post-production information
The output of this review is the Risk Management Report — a document summarizing the risk management process, confirming that the plan was followed, and stating the overall residual risk conclusion. The risk management report is one of the documents Notified Bodies specifically request and review during technical documentation assessments.
Step 8 — Production and Post-Production Information (Clause 10)
A substantial change in ISO 14971:2019 is the expansion of requirements for production and post-production activities. Manufacturers must perform a full review of the risk management process prior to commercial distribution — ensuring that the risk management plan has been appropriately implemented, the overall risk is acceptable, and that procedures are in place to gather and maintain risk data during production and post-production.
Post-production monitoring must cover: complaint data and vigilance reports; field feedback from users, healthcare professionals, and service engineers; published scientific and clinical literature on the device or similar devices; changes in clinical practice or state of the art; and post-market surveillance data from EU MDR post-market surveillance requirements.
Information collected should include any newly identified hazards, changes that affect risk analysis calculations, and results of regular reviews of the risk management file. ISO 14971 aligns closely with the ISO 13485:2016 Section 8 requirements for feedback, analysis of data, and CAPA.
This post-production loop is what makes ISO 14971 a living process rather than a one-time documentation exercise — the risk management file must be updated throughout the device’s commercial lifetime as new information becomes available.
COMPLETE CATALOG
Find the documentation you need — instantly.
Whether you need a complete kit or just one specific SOP, our catalog has it. 45 process packages and 3 complete bundles, all instantly downloadable and fully editable.
- ✓Complete bundles or individual packages
- ✓45 process packages from €69 each
- ✓ISO 13485 · MDSAP · Combined Kit
The Risk Management File — What Must Be in It
ISO 14971 Section 4.5 states that “the risk management file shall provide traceability for each identified hazard.” Traceability means the manufacturer can locate all records and documents applicable to risk management — facilitating the risk management process and enabling more efficient auditing.
The risk management file is not a single document — it is a structured collection of records that together demonstrate the complete risk management process. At minimum it must contain:
Risk Management Plan — device-specific, covering scope, criteria, responsibilities, and methods
Hazard and hazardous situation documentation — complete list of identified hazards and the sequences of events leading to hazardous situations
Risk estimation records — for each hazardous situation: probability estimate, severity level, and resulting risk level
Risk evaluation records — the acceptability decision for each estimated risk, with reference to the criteria in the plan
Risk controls documentation — description of each control measure implemented, implementation verification evidence, and effectiveness verification evidence
New hazards from risk controls — documentation that risk controls were assessed for introduction of new hazards
Overall residual risk evaluation — the benefit-risk analysis and overall residual risk conclusion
Risk Management Report — the formal pre-market review confirming plan implementation and overall risk acceptability
Post-production information records — evidence of ongoing monitoring, data sources reviewed, and any updates to the risk management file triggered by post-market findings
The Risk Management File shall be kept up-to-date throughout the product lifecycle, until the last product in the field is removed from use and properly disposed of. The entire RMF may only be destroyed following Document Control requirements after a device is no longer available for use.

Figure 3 — Risk management file structure and content
ISO 14971 and Its Relationship to Other Standards
ISO 14971 does not operate in isolation — it is explicitly referenced and required by the majority of other medical device standards and regulations. Understanding these relationships is essential for building an integrated compliance system rather than treating risk management as a standalone exercise.
ISO 13485:2016: The ISO 13485 quality management system requires that risk management be applied throughout the entire QMS — in design controls, production process control, supplier evaluation, complaint handling, CAPA, and management review. ISO 13485 Clause 7.1 explicitly requires risk management activities as part of product realization planning. The two standards are complementary — ISO 13485 defines the QMS framework, ISO 14971 defines the product risk management process within it.
EU MDR 2017/745: EN ISO 14971:2019 is harmonized with EU MDR and IVDR. The EU MDR technical documentation requirements under Annex II Section 4 require a complete risk management file per ISO 14971, a benefit-risk analysis demonstrating that benefits outweigh residual risks, and integration of post-market surveillance data into the risk management file continuously. EU MDR also requires that all risks be reduced as far as possible — a stricter standard than the base ISO 14971 acceptability threshold approach.
IEC 62304: For medical device software, IEC 62304 defines the software lifecycle processes. It uses ISO 14971 as its risk management reference — the software safety class (A, B, or C) assigned to each software item under IEC 62304 is determined by the severity of harm that could result from software failure, as defined in the ISO 14971 hazard analysis. Software hazards identified through the ISO 14971 process feed directly into the IEC 62304 software development requirements. The SOUP management process under IEC 62304 also generates inputs to the ISO 14971 risk analysis — anomalies in third-party software components must be evaluated for their impact on risk.
IEC 62366 (Usability Engineering): Usability engineering and risk management are deeply interconnected in ISO 14971. Use errors identified during usability evaluation are hazards that must be processed through the risk management framework. Conversely, risk controls implemented as information for safety (IFS) — warnings, precautions, training requirements — must be validated through usability testing as part of effectiveness verification.
IEC 81001-5-1 (Cybersecurity): Cybersecurity risks must be integrated into the ISO 14971 risk management process as a distinct category. IEC 81001-5-1 is a standard focused on cybersecurity risk management related to medical devices and software. Scalehealth The cybersecurity threat model — threat identification, vulnerability assessment, risk estimation — feeds into the ISO 14971 risk register, and cybersecurity controls are implemented and verified within the ISO 14971 framework. Our guide to medical device cybersecurity testing covers how security testing integrates with risk verification.
ISO 10993-1: Biocompatibility evaluation is explicitly part of the ISO 14971 risk management process. The ISO 10993-1:2025 biological evaluation process generates risk information about biological hazards — cytotoxicity, sensitization, systemic toxicity — that must be integrated into the risk management file.
MDSAP: For manufacturers pursuing MDSAP certification, ISO 14971 risk management is audited as part of the single audit that covers all five participating regulatory authorities. MDSAP auditors assess risk management integration across design controls, production, and post-market activities.
Common Notified Body and FDA Findings in ISO 14971 Audits

Figure 4 — Most common ISO 14971 audit findings
Generic or non-objective acceptability criteria is one of the most consistently cited findings in Notified Body technical documentation reviews. Many risk management plans define acceptability criteria in vague terms such as “all risks shall be reduced to acceptable levels” or reference ALARP without defining objective thresholds. The risk management plan must define specific, measurable criteria — a severity scale with clinical anchors, a probability scale with quantitative or qualitative definitions, and a risk matrix with clearly delineated zones. Without objective criteria, the entire risk evaluation process is unverifiable.
Risk management treated as a silo — the most operationally damaging finding. ISO 14971 is not an FMEA and not a one-time deliverable — it is a closed-loop system that ties design inputs, verification and validation, production controls, and post-market signals into a single living Risk Management File. Risk management files that are completed during design and never updated after market release consistently receive major findings. The file must demonstrate active integration with the CAPA system, complaint analysis, and post-market surveillance data.
Inadequate hazard identification — particularly the absence of reasonably foreseeable misuse scenarios, software-specific hazards, cybersecurity hazards, and use error hazards. If you are using FMEA only for your risk management efforts, you are not meeting the intent of ISO 14971. FMEA is a valuable tool for process and design risk analysis but is not sufficient as the sole hazard identification method — preliminary hazard analysis, fault tree analysis, and use scenario analysis must also be applied as appropriate.
No effectiveness verification for risk controls — risk controls are listed in the risk management file but there is no documented evidence that they were tested, validated, or confirmed to achieve the intended risk reduction. Every risk control must have associated objective evidence — test reports, clinical data, analysis results, or validated usability study outcomes — demonstrating that it works as intended.
Missing benefit-risk analysis and overall residual risk evaluation — the Clause 8 evaluation is absent or superficial. For EU MDR-covered devices this is particularly critical because the EU MDR technical documentation under Annex II explicitly requires a benefit-risk determination supported by clinical evidence. The overall residual risk evaluation must be a documented, explicit conclusion — not implied by the absence of red risks in the risk matrix.
Incomplete traceability — the risk management file cannot demonstrate the complete chain from hazard identification through risk estimation, evaluation, control implementation, effectiveness verification, and residual risk recording for each hazard. ISO 14971 Section 4.5 requires that the risk management file provide traceability for each identified hazard — the manufacturer must be able to locate all records applicable to every hazard. A traceability matrix that links each hazard to its hazardous situations, risk controls, verification evidence, and residual risk record is the most efficient way to demonstrate this.
ISO 14971 Risk Analysis Tools — Choosing the Right Method
ISO 14971 does not mandate a specific risk analysis tool. Manufacturers must select the methods most appropriate to the device type, complexity, and phase of development. The most widely used tools in the medical device industry are:
Failure Mode and Effects Analysis (FMEA): Analyzes potential failure modes of device components or processes, their causes, and their effects on device function and patient safety. FMEA is the most widely used risk analysis tool in medical device development but must be used appropriately — it is most effective for analyzing component failures and manufacturing process risks, not for comprehensively identifying all device hazards. Design FMEA (dFMEA) and Process FMEA (pFMEA) serve different purposes and both may be required.
Fault Tree Analysis (FTA): A top-down deductive analysis that models how a hazardous situation or harm event can occur, working backwards from the undesired outcome to identify the combination of component failures or events that could produce it. FTA is particularly valuable for complex devices with multiple interacting subsystems and for analyzing low-probability but high-severity scenarios. It complements FMEA — FTA shows how failures combine, while FMEA shows what can fail individually.
Preliminary Hazard Analysis (PHA): A structured brainstorming technique typically applied early in the design phase — before detailed design is completed — to identify potential hazards associated with the device concept. PHA generates inputs to the more detailed FMEA and FTA analyses that follow.
Hazard and Operability Study (HAZOP): Originally developed for chemical processes, HAZOP is increasingly applied to medical device software and systems. It uses structured guidewords (too much, too little, early, late, wrong direction, etc.) to identify deviations from intended function and their potential safety consequences.
Use Error Analysis (aligned with IEC 62366): Systematic analysis of use scenarios and potential use errors — misconnections, incorrect settings, misinterpretation of outputs — that represent a significant category of hazards for many device types. Use error analysis feeds directly into the ISO 14971 risk management process and connects to the usability engineering requirements of IEC 62366.
The practical recommendation is to use a combination of methods — PHA for early-stage hazard identification, FMEA for design and process analysis, FTA for complex failure scenarios, and use error analysis for human factors hazards. The choice and rationale for each method should be documented in the risk management plan.
Practical Guide to Building Your ISO 14971 Risk Management File
For manufacturers building a new risk management file or upgrading an existing one to ISO 14971:2019, the following sequence reflects industry best practice.
Start with the risk management plan before any design work begins. The plan is not a retrospective document — it should define the risk management approach before the design inputs are finalized. This ensures that the device characteristics documented in the design history file are aligned from the start with the hazard categories and acceptability criteria in the risk management plan.
Define your risk acceptability criteria with clinical specificity. Generic severity descriptions (“minor,” “serious,” “critical”) without clinical anchors produce inconsistent risk estimations across reviewers. Define each severity level with clinical examples relevant to the intended patient population and indication — “serious” for a patient monitoring device might be defined as “harm requiring hospitalization or significant medical intervention,” with examples from the clinical literature.
Document the intended use and reasonably foreseeable misuse comprehensively. A narrow intended use definition reduces the apparent scope of hazards — but Notified Bodies will challenge any intended use that appears to have been written to minimize the hazard identification burden. Document all foreseeable use scenarios including off-label use, pediatric use where relevant, use by lay persons if applicable, and use in resource-limited settings if the device is intended for global markets.
Use Annex C of ISO 14971:2019 as your hazard identification checklist. Annex C provides a catalogue of hazard categories — energy hazards, biological hazards, environmental hazards, hazards related to use, information hazards — that should be systematically reviewed for applicability to the specific device. Mark each category as applicable or not applicable, with justification for non-applicable items. This demonstrates systematic coverage to Notified Body reviewers.
Maintain a living risk register — not a static spreadsheet. Risk management is not one document, one audit, or one design review — it is the outcome of a mature quality management system that consistently produces the right evidence. LinkedIn Establish a change control process that triggers risk management file review whenever the device design, manufacturing process, intended use, or post-market data changes. The risk register must be accessible to design, clinical, quality, and regulatory functions — not locked in the quality department.
Build traceability into the structure from the start. Assign a unique identifier to each hazard when it is identified, and use that identifier to link all associated records — risk estimation, evaluation decision, risk controls, implementation verification, effectiveness verification, and residual risk — across the entire file. This makes audit preparation significantly faster and prevents the most common traceability finding.
Conduct the risk management review before any Notified Body submission. The risk management review is a formal gate — it must confirm that the plan has been fully implemented, all identified hazards have been processed through the complete risk management cycle, all risk controls have been verified, and the overall residual risk is acceptable. For EU MDR submissions, the risk management review must also confirm that the benefit-risk analysis is supported by the clinical evidence in the clinical evaluation report.
✦ BUNDLE · RISK + CLINICAL
The clinical and risk file for EU MDR — designed to reference each other.
The Risk Management Kit and the EU MDR Clinical Documentation Kit, bundled together. The Risk Management Report cross-references the CER for benefit-risk conclusions, and the GSPR Checklist references both clinical and risk evidence for each requirement.
- ✓14 templates · risk + clinical, cross-referenced
- ✓Save €149 vs buying the kits separately
ISO 14971 and the State of the Art
One of the most practically important — and most often underestimated — concepts in ISO 14971:2019 is the state of the art. The 2019 edition introduced a formal definition: the state of the art is the developed stage of technical capability at a given time as regards products, processes and services, based on the relevant consolidated findings of science, technology, and experience.
The state of the art is relevant at two points in the ISO 14971 process:
Risk acceptability assessment: A risk that is inherent to all devices of the same type and cannot be eliminated with available technology may be acceptable based on the state of the art, even if it exceeds the manufacturer’s internal acceptability thresholds. If all implantable cardiac devices on the market carry a certain rate of lead dislodgement, and the current state of the art cannot eliminate this risk, a manufacturer must still minimize it as far as possible — but cannot be held to a standard that no device currently meets.
Overall residual risk evaluation: The overall residual risk of the device must be evaluated against the clinical benefits in the context of the state of the art — meaning in comparison to available alternatives and current clinical practice. A higher overall residual risk may be acceptable if there is no alternative treatment and the clinical benefit is substantial.
In practice, the state of the art must be documented with references — clinical literature, regulatory guidance, international standards, and published data from similar devices. A state of the art assessment that cites no external evidence will not withstand scrutiny from a Notified Body reviewer.
Frequently Asked Questions
Is ISO 14971 mandatory for all medical device manufacturers? ISO 14971 is not legally mandatory in the sense that compliance with the standard itself is required — but the regulatory frameworks that are legally mandatory (EU MDR, FDA QMSR, ISO 13485) all require an equivalent risk management process and recognize ISO 14971 as the internationally accepted method. In practice, demonstrating compliance with ISO 14971 is the universally expected approach — without an ISO 14971-aligned risk management process, demonstrating compliance with EU MDR, FDA QMSR, ISO 13485, or any other major regulatory framework is extremely difficult.
What is the difference between the risk management file and the risk management report? The risk management file is the complete collection of all risk management records — the plan, hazard list, risk estimates, evaluation decisions, risk controls documentation, benefit-risk analysis, and post-production monitoring records. The risk management report is a specific document within the file — the formal pre-market review output that summarizes the risk management process, confirms the plan was implemented, and states the overall residual risk conclusion. The report is a snapshot; the file is the ongoing record.
How often must the risk management file be updated? The risk management file must be updated whenever there is a change that could affect risk — design changes, manufacturing process changes, new clinical evidence, post-market surveillance findings, CAPA outputs, or changes in applicable standards or the state of the art. In addition, the risk management review should be conducted before any significant submission or regulatory interaction, and post-production monitoring requires systematic periodic review of field data. There is no minimum prescribed frequency in the standard — the appropriate frequency depends on the risk profile of the device and the pace of post-market information generation.
Does ISO 14971 apply to software as a medical device (SaMD)? Yes — ISO 14971:2019 explicitly includes software as a medical device and in vitro diagnostic medical devices within its scope, to avoid any misunderstanding that these devices might be excluded. For SaMD, ISO 14971 provides the risk management framework, while IEC 62304 defines the software lifecycle processes. The two standards work together — the software safety class assigned under IEC 62304 is determined by the severity of harm from the ISO 14971 hazard analysis, and software anomalies identified during development or post-market must be evaluated through the ISO 14971 risk process.
What is the relationship between ISO 14971 and the EU MDR benefit-risk analysis? EU MDR requires that the technical documentation include a benefit-risk analysis demonstrating that benefits outweigh risks. This requirement maps directly to ISO 14971 Clause 8 — the overall residual risk evaluation and benefit-risk analysis. The difference is that EU MDR requires this analysis to be supported by clinical evidence from the clinical evaluation. Our detailed article on benefit-risk analysis under EU MDR covers this intersection in depth.
What is ISO/TR 24971 and do I need it? ISO/TR 24971:2020 is the companion technical report to ISO 14971:2019. It is not normative — it contains no requirements — but it provides detailed implementation guidance, worked examples, and clarifications that make the standard significantly more practical to apply. It covers topics including risk acceptability criteria definition, probability estimation methods, risk analysis tools, benefit-risk analysis methodology, and post-production monitoring. Any manufacturer implementing or upgrading their ISO 14971 process should treat ISO/TR 24971:2020 as an essential reference alongside the standard itself.
Conclusions
ISO 14971 risk management is not a documentation exercise — it is the safety backbone of medical device development. When run properly, it drives safer designs, cleaner files, and faster, defensible decisions when things go wrong. When treated as a retrospective paperwork activity, it creates a file that satisfies no one — not the manufacturer, not the Notified Body, and most importantly not the patients who depend on the device being as safe as it can reasonably be.
The key principles that define a mature ISO 14971 process are: continuity — risk management must be active from first concept to last device in the field; integration — the risk management file must connect to design controls, production, CAPA, and post-market surveillance, not exist as a standalone document; traceability — every identified hazard must be traceable through estimation, evaluation, control, verification, and residual risk documentation; and proportionality — the depth and rigor of risk management must match the risk profile of the device.
For manufacturers building or rebuilding their risk management system, the right documentation framework is the essential starting point. The ISO 14971 Risk Management SOP package on MD Regulatory includes a complete risk management procedure aligned with ISO 14971:2019, a risk management plan template, risk register with traceability matrix, risk acceptability criteria template, benefit-risk analysis form, and risk management report template — all written to current Notified Body expectations and deployable within an existing ISO 13485 QMS.
This article is part of the MD Regulatory risk management series. Related articles: Benefit-Risk Analysis under EU MDR· EU MDR Technical Documentation Requirements · ISO 13485 Complete Guide · IEC 62304 Medical Device Software · ISO 13485 CAPA Procedure · Medical Device Cybersecurity Testing






3 Comments
Comments are closed.