Biocompatibility Testing for Medical Devices: ISO 10993 Complete Guide
Table of Contents
Biocompatibility testing is one of the most fundamental requirements in medical device development. Any device that comes into contact with the human body — whether it touches intact skin, penetrates mucosal membranes, contacts blood, or is implanted permanently in tissue — must demonstrate that its materials do not cause unacceptable biological harm to patients or users.
For the purpose of the ISO 10993 family of standards, biocompatibility is defined as the ability of a medical device or material to perform with an appropriate host response in a specific application. This definition captures something fundamental: biocompatibility is not a material property in isolation — it depends on the specific context of clinical use, the contact type, the duration of exposure, and the patient population. ISO
ISO 10993-1 defines the principles and requirements for assessing a device’s biological safety within the broader risk management framework established by ISO 14971. The standard guides manufacturers and evaluators through the process of identifying, assessing, and managing biological risks associated with materials, design choices, and tissue contact during a device’s intended use. Semrush
This guide covers the complete biocompatibility testing framework — from the ISO 10993 series structure through biological evaluation planning, device categorization, test selection, chemical characterization, and the key changes introduced by ISO 10993-1:2025. For context on how biocompatibility integrates into the broader ISO 14971 risk management process, refer to our complete risk management guide.
✦ EU MDR CLINICAL DOCUMENTATION KIT
Clinical evaluation, PMS, PMCF and SSCP — covered end-to-end.
8 Word templates for the full clinical lifecycle under EU MDR — CEP, CER, CDP, PMS Plan, PSUR, PMCF Plan and Evaluation Report, plus SSCP for Class III and implantables. Aligned with MDCG 2020-5/6/7/8/13 and 2022-21.
- ✓8 Word templates · pre-market + post-market
- ✓MDCG-aligned · ready for Notified Body submission
What Is the ISO 10993 Series?
The ISO 10993 series is a family of international standards developed by ISO Technical Committee 194 (ISO/TC 194) that provides the globally recognized framework for biological evaluation of medical devices. The ISO 10993 series provides guidelines for assessing the potential risks associated with the use of medical devices, with each standard covering a specific aspect of biocompatibility.
The series currently comprises 23 active parts — each addressing a specific biological endpoint or aspect of the evaluation process:
ISO 10993-1:2025 — The foundational standard. Defines the principles, requirements, and process for biological evaluation within a risk management framework. All other parts of the series operate under the framework established by Part 1.
ISO 10993-3:2014 — Tests for genotoxicity, carcinogenicity, and reproductive toxicity
ISO 10993-4:2017 — Selection of tests for interactions with blood
ISO 10993-5:2009 — Tests for in vitro cytotoxicity
ISO 10993-6:2016 — Tests for local effects after tissue contact (formerly “after implantation”)
ISO 10993-7:2008 — Ethylene oxide sterilization residuals
ISO 10993-9:2019 — Framework for identification and quantification of potential degradation products
ISO 10993-10:2021 — Tests for skin sensitization
ISO 10993-12:2021 — Sample preparation and reference materials
ISO 10993-13:2010 — Degradation products from polymeric medical devices
ISO 10993-15:2019 — Degradation products from metals and alloys
ISO 10993-17:2023 — Toxicological risk assessment of medical device constituents
ISO 10993-18:2020 — Chemical characterization of medical device materials within a risk management process
ISO 10993-22:2017 — Guidance on nanomaterials
ISO 10993-23:2021 — Tests for irritation
ISO 10993-1 is the only normative framework standard — the others define specific testing methodologies referenced by ISO 10993-1 when particular biological endpoints require evaluation.

Figure 1 — ISO 10993 biological evaluation framework overview
ISO 10993-1:2025 — What Changed and Why It Matters
The 2025 edition of ISO 10993-1 is the sixth edition and represents the most significant revision since the 2009 update. The sixth edition has been completely reorganized to align with ISO 14971. It adds new guidance on exposure duration, materials characterization, and identification of biological hazards. Terminology has been updated for clarity.
For manufacturers with existing biological evaluation documentation based on ISO 10993-1:2018, a gap analysis against the 2025 edition is now recommended — particularly for EU market where quick adoption is expected as the revision is recognized as state of the art.
Key changes in ISO 10993-1:2025:
New four-category device classification: In the previous version, medical devices were categorized as surface, externally communicating, or implantable, in addition to their contact-site-specific designation. In the newest version, devices are categorized only based on contact type: intact skin, intact mucosal membranes, breached or compromised surfaces of internal tissues other than blood, and blood. This simplification reduces ambiguity in device categorization while maintaining the exposure duration framework (limited, prolonged, long-term).
Biological equivalence formalized: The revised standard introduces and formalizes the concept of biological equivalence. Biological equivalence may be claimed when the chemical and physical characteristics and manufacturing processes of a medical device or material under evaluation are sufficiently similar to those of a comparator device or material with established biological safety. Any differences between the device and the comparator must be demonstrated not to introduce new or increased biological risks.
Mandatory gap analysis: The revised standard introduces a clear expectation for the performance of a gap analysis early in the biological evaluation process. This must be documented before decisions about testing are made.
Expanded biological effects scope: One prominent change is the addition of two new biological effects: neurotoxicity and immunotoxicity, added in response to emerging information regarding novel device materials and their interactions with the human body.
CMR and endocrine disruptors: The standard adds CMR (carcinogenic, mutagenic, reproductive toxicity) and endocrine disruptor assessment into scope. Novel and complex materials, including nanomaterials, require deeper characterization and chemical data.
Exposure duration redefined: Exposure duration is now defined by total days of clinical use rather than cumulative sessions. This change may result in re-classification of exposure categories and trigger more comprehensive hazard testing.
Reduced animal testing emphasis: Animal tests should not be performed unless no suitable alternative methods are available and unless they are essential to demonstrate biological safety. Chemical characterization and in vitro methods should be exhausted before in vivo testing is considered.
Device Categorization Under ISO 10993-1:2025
Before selecting biocompatibility tests, every device must be categorized based on two dimensions: contact type and contact duration. These two dimensions together determine the minimum biological endpoints that must be evaluated.
Contact Type (ISO 10993-1:2025 Table A.1)
The 2025 edition simplifies device categorization to four contact categories:
Intact skin: Devices that contact only the unbroken surface of the skin — adhesive bandages, ECG electrodes, blood pressure cuffs, external prostheses.
Intact mucosal membranes: Devices contacting mucosal surfaces such as the oral cavity, nasal passages, gastrointestinal tract, urogenital tract — dental impression materials, urinary catheters, gastrointestinal endoscopes.
Breached or compromised surfaces / internal tissues other than blood: Devices contacting wounded or compromised skin, or internal tissues not in contact with the circulatory system — wound dressings, surgical instruments, orthopaedic implants in bone or soft tissue.
Blood: Devices contacting the cardiovascular system or blood — cardiac catheters, vascular grafts, heart valves, dialysis membranes, blood oxygenators.
Contact Duration
Three duration categories apply regardless of contact type:
Limited contact: Total contact duration of 24 hours or less
Prolonged contact: Total contact duration between 24 hours and 30 days
Long-term contact: Total contact duration exceeding 30 days
An important nuance introduced in the 2025 edition: exposure duration is now defined by total days of clinical use rather than cumulative sessions. This means that a device used repeatedly for short periods may be reclassified as prolonged or long-term based on accumulated total exposure — triggering more comprehensive biological evaluation requirements.
The Biological Evaluation Process — Step by Step
Step 1 — Device Characterization
The biological evaluation process begins with a comprehensive characterization of the device — not the materials in isolation, but the final finished device as it will be used clinically. This includes: all materials and components in contact with the body or potentially in contact through indirect pathways; all manufacturing processes and process aids (lubricants, mold release agents, cleaning agents); sterilization method and any sterilization residuals; the intended use environment; and the patient population including any vulnerable groups.
Crucially, reasonably foreseeable misuse scenarios must now be explicitly considered in the exposure assessment under ISO 10993-1:2025 — consistent with the ISO 14971 approach to hazard identification covered in our ISO 14971 risk management guide.
Step 2 — Biological Evaluation Plan (BEP)
The Biological Evaluation Plan is the strategic document that defines how the biological evaluation will be conducted for a specific device. It is the biocompatibility equivalent of the risk management plan under ISO 14971 — device-specific, documented before testing begins, and subject to review and update throughout the device lifecycle.
The BEP must include: the device categorization (contact type and duration); a review of all existing biological safety data for the device materials; a gap analysis identifying what additional data is needed; the proposed strategy for addressing data gaps — through chemical characterization, toxicological risk assessment, biological equivalence, or testing; and the biological endpoints to be evaluated.
Documentation must clearly demonstrate how biological hazards were identified, evaluated, and controlled, and how these activities integrate with the overall risk management process.
Step 3 — Chemical Characterization (ISO 10993-18)
Chemical characterization is now the expected first step after the BEP — before any biological testing is considered. Chemical characterization remains as an important tool to use in identifying biological hazards, biologically hazardous situations, and biological risks associated with a final finished device, its materials, and manufacturing processes.
Chemical characterization under ISO 10993-18 includes: identification of all chemical constituents of the device and its materials; extractables and leachables testing — the chemical compounds that can be released from the device under simulated or actual use conditions; an assessment of CMR substances and endocrine disruptors (new in the 2025 edition); and characterization of degradation products using the appropriate ISO 10993 parts (10993-9, -13, -15, -16).
The output of chemical characterization feeds directly into the toxicological risk assessment — and may be sufficient to conclude on biological safety for many endpoints without requiring in vivo animal testing.
Step 4 — Toxicological Risk Assessment (ISO 10993-17)
The toxicological risk assessment (TRA) evaluates whether the chemical compounds identified through characterization pose an unacceptable biological risk at the expected patient exposure levels. It compares the actual or estimated patient exposure to each chemical against established toxicological thresholds — tolerable daily intake (TDI), tolerable contact levels (TCL), or other appropriate limits from regulatory databases (EFSA, ECHA, WHO, FDA).
Where chemical characterization and TRA together demonstrate that biological risks are acceptable, additional in vitro or in vivo testing may not be required for specific endpoints. This approach — which the 2025 edition explicitly supports — can significantly reduce testing burden, timelines, and animal use.
COMPLETE CATALOG
Find the documentation you need — instantly.
Whether you need a complete kit or just one specific SOP, our catalog has it. 45 process packages and 3 complete bundles, all instantly downloadable and fully editable.
- ✓Complete bundles or individual packages
- ✓45 process packages from €69 each
- ✓ISO 13485 · MDSAP · Combined Kit
Step 5 — Biological Testing (When Required)
Biological testing is conducted when chemical characterization and TRA are insufficient to conclude on biological safety for specific endpoints. Test selection must be justified based on the device category, contact duration, and the specific biological endpoints that cannot be addressed through chemical data alone.
Cytotoxicity, irritation, and sensitization are the “Big Three” assessments that must be performed for almost all medical devices being introduced to the market — depending on the type of device and its intended use, additional tests may be required.
The biological endpoints defined in ISO 10993-1 and addressed through specific parts of the series include: cytotoxicity (ISO 10993-5), sensitization (ISO 10993-10), irritation (ISO 10993-23), systemic toxicity — acute and subacute (ISO 10993-11), genotoxicity (ISO 10993-3), haemocompatibility (ISO 10993-4), implantation/local tissue effects (ISO 10993-6), chronic toxicity (ISO 10993-11), carcinogenicity (ISO 10993-3), reproductive and developmental toxicity (ISO 10993-3), and — new in the 2025 edition — neurotoxicity and immunotoxicity.
Step 6 — Biological Evaluation Report (BER)
The Biological Evaluation Report is the final output of the biocompatibility evaluation — a comprehensive document summarizing all data reviewed, all testing conducted, the conclusions reached, and the overall biological risk determination. The BER integrates with the ISO 14971 risk management file and forms part of the EU MDR technical documentation under Annex II.
The BER is a living document — it must be updated whenever there are changes to device materials, manufacturing processes, or post-market biological safety signals.
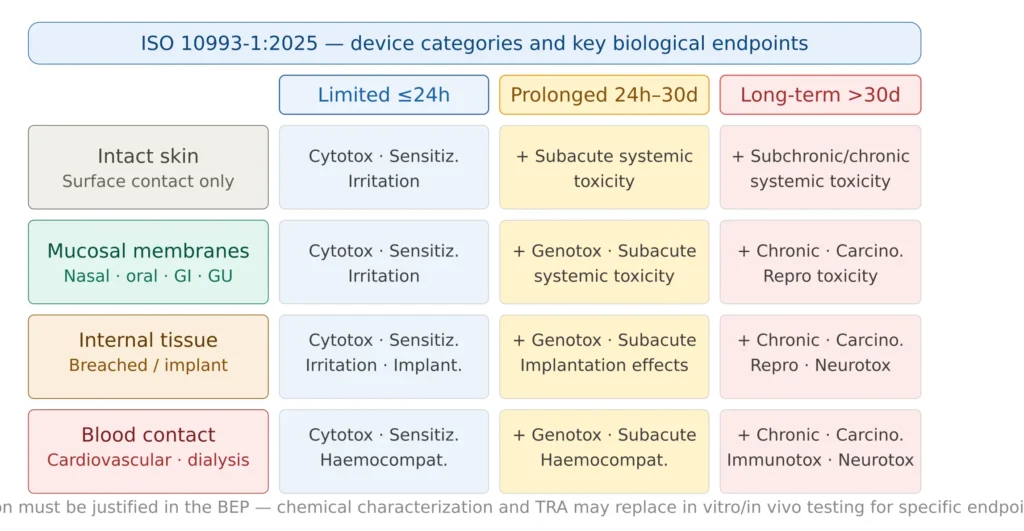
Figure 2 — Device contact categories and required biological endpoints
The Three Most Important Biocompatibility Tests
While the full biocompatibility testing program can include up to 15+ endpoints for long-term implants, three tests are required for virtually every medical device regardless of contact type and duration.
Cytotoxicity Testing (ISO 10993-5)
Cytotoxicity testing evaluates whether device materials or their extracts are toxic to living cells. It is the most fundamental biocompatibility screen — the first test performed and the one most likely to detect gross material toxicity early in development.
ISO 10993-5 defines three principal methods: direct contact (material placed directly on cell culture), indirect contact (material extract applied to cells), and elution/extract method (extract prepared per ISO 10993-12 applied to the culture). The L929 mouse fibroblast cell line is the most widely used for cytotoxicity evaluation.
Cytotoxicity testing is fast, cost-effective, and can be performed early in the design process — making it the ideal first biological screen before more expensive or time-consuming tests are commissioned.
Sensitization Testing (ISO 10993-10)
Sensitization testing evaluates whether device materials can cause an allergic reaction (delayed hypersensitivity) following repeated or prolonged exposure. This is particularly important for devices containing latex, certain metals (nickel, chromium, cobalt), and adhesives.
ISO 10993-10 defines in vivo sensitization tests — the guinea pig maximization test and the Buehler test. In vitro alternatives are under development but the FDA does not currently consider in vitro methods as equivalent for sensitization — meaning in vivo testing is still typically required for US submissions.
Irritation Testing (ISO 10993-23)
ISO 10993-23 covers tests for irritation — the local tissue response to device contact that does not involve immune sensitization. It replaced the older ISO 10993-10 irritation provisions and aligns more strongly with a risk-based approach, encouraging the use of in vitro methods and chemical data before in vivo testing.
Biocompatibility Testing and EU MDR Requirements
EU MDR 2017/745 does not specify biocompatibility tests directly — but it requires, through the General Safety and Performance Requirements (GSPRs) of Annex I, that devices are designed and manufactured to be safe for their intended use, and that biological risks are eliminated or reduced as far as possible.
EN ISO 10993-1:2025 is recognized as the state of the art for biological evaluation under EU MDR. As part of the EU MDR technical documentation under Annex II, the biological evaluation documentation must include the BEP, all testing and characterization reports, and the BER — all integrated into the ISO 14971 risk management file.
For Class III implantable devices, Notified Bodies apply particularly close scrutiny to biological evaluation data — especially where novel materials are used, where chemical characterization data is incomplete, or where the TRA relies on tolerances derived from food or pharmaceutical contexts rather than medical device-specific databases.
The connection between biocompatibility and ISO 13485 is also direct — ISO 13485 Clause 7.3 (design and development) requires that biological safety be addressed in design inputs and design verification/validation. The BEP should be initiated at the design input stage, and the BER serves as part of the design verification evidence.
Biocompatibility Testing and FDA Requirements
The FDA has developed guidance to assist industry in preparing PMAs, 510(k)s, and De Novo requests for medical devices that come into direct or indirect contact with the human body — to determine the potential for an unacceptable adverse biological response resulting from contact of the component materials of the device with the body.
The FDA’s approach to biocompatibility is defined in its guidance document “Use of International Standard ISO 10993-1” and in the FDA-modified testing matrix, which modifies the ISO 10993-1 test selection table for certain device categories and endpoints. Key differences between FDA and ISO/EU expectations include:
Chemical characterization: The FDA strongly encourages — and in practice expects — chemical characterization as the first step, consistent with ISO 10993-18. The FDA guidance explicitly states that a risk-based approach using chemical data should be used to determine whether in vivo testing is necessary.
Sensitization: As noted, the FDA does not currently accept in vitro sensitization alternatives as equivalent to guinea pig tests — making in vivo sensitization testing generally required for US submissions even when in vitro data exists.
Genotoxicity: Under the revised ISO 10993-1, genotoxicity evaluation is now applicable to all medical devices with prolonged contact, with the exception of devices that contact intact skin only. The FDA’s approach aligns closely with this expanded scope.
Extractables and leachables: The FDA expects chemical characterization of extractables and leachables for devices with prolonged or long-term contact, consistent with ISO 10993-18. Analytical chemistry reports with identified compounds and quantification against toxicological thresholds are standard submission content.

Figure 3 — Common biocompatibility testing audit findings
Practical Guidance — Building Your Biocompatibility Strategy
Start at design input — not at design verification. The most costly biocompatibility mistakes happen when manufacturers select materials without early biological evaluation input, discover a biocompatibility issue at the verification stage, and face an expensive redesign. Material selection decisions made with biocompatibility in mind from the start — choosing materials with established biological safety profiles and avoiding novel compounds without characterization data — dramatically reduce downstream testing burden.
Use chemical characterization data strategically. A comprehensive chemical characterization report, when coupled with a well-executed TRA, can eliminate the need for several in vivo biological tests. This is not a shortcut — it is the scientifically rigorous approach explicitly endorsed by ISO 10993-1:2025 and the FDA. The investment in good chemical characterization at the start pays for itself many times over in avoided testing costs, animal use, and time.
Understand the difference between extractables and leachables. Extractables are the chemical compounds that can be released from device materials under aggressive laboratory conditions — they define the worst-case exposure ceiling. Leachables are the compounds actually released under normal or simulated use conditions — they define the realistic patient exposure. Both must be characterized and assessed; the distinction affects how the TRA is conducted and what toxicological thresholds apply.
Document biological equivalence rigorously when claiming it. Biological equivalence is a powerful tool for avoiding redundant testing when a new device uses the same materials as a predicate with established biological safety. But the equivalence claim must be fully documented — demonstrating that the chemical composition, manufacturing processes, and contact conditions are sufficiently similar, and that any differences do not introduce new biological risks. A poorly documented equivalence claim is a common source of Notified Body questions.
Maintain the BER as a living document. Any change to device materials, component suppliers, manufacturing processes, sterilization method, or intended use must trigger a review of the biological evaluation. The BER must be updated to reflect the impact of the change — even if the conclusion remains that no additional testing is required. An outdated BER is a consistent finding in Notified Body surveillance audits.
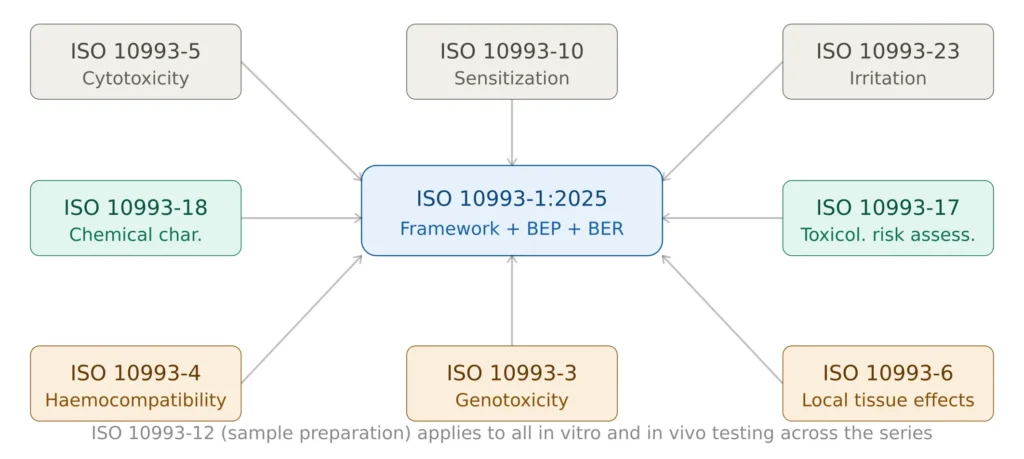
Figure 4 — ISO 10993 series key parts and their role in the biocompatibility program
Frequently Asked Questions
Does every medical device need biocompatibility testing? Every device that contacts the human body requires a biological evaluation — but biological evaluation does not always mean in vitro or in vivo testing. For many devices, particularly those made from well-characterized materials with established safety profiles, chemical characterization and a documented TRA may be sufficient to conclude on biological safety without any laboratory testing. The BEP documents this determination.
What is the difference between a BEP and a BER? The Biological Evaluation Plan (BEP) is a prospective document — written at the start of the evaluation, it defines the strategy: what data exists, what gaps exist, and how those gaps will be addressed. The Biological Evaluation Report (BER) is the retrospective output — it summarizes all data collected, all analyses performed, and the overall biological risk conclusion. Both are required; neither replaces the other.
When should biocompatibility evaluation begin in the design process? As early as possible — ideally at the design input stage when materials are first being selected. Early engagement with biocompatibility requirements prevents costly material changes at the design verification stage, enables informed supplier selection, and allows chemical characterization to proceed in parallel with other development activities.
How does ISO 10993-1:2025 differ from ISO 10993-1:2018? The key changes are: simplified four-category device classification; formalization of biological equivalence; mandatory gap analysis before testing decisions; expanded biological effects scope including neurotoxicity and immunotoxicity; addition of CMR and endocrine disruptor assessment; exposure duration redefined by total clinical use days rather than cumulative sessions; and stronger integration with ISO 14971 risk management terminology and framework. For manufacturers with existing 2018-based evaluations, a gap analysis against the 2025 edition is recommended.
Is ISO 10993-1:2025 recognized by the FDA? As of April 2026, the FDA had not yet formally recognized ISO 10993-1:2025 in its Recognized Consensus Standards database — the FDA typically takes 12-24 months to recognize new editions of international standards. Manufacturers preparing US submissions should monitor the FDA Recognized Consensus Standards database for the recognition status. For EU submissions, ISO 10993-1:2025 is recognized as state of the art and its application is expected.
How does biocompatibility integrate with the ISO 14971 risk management file? The BER is part of the ISO 14971 risk management file — biological hazards are a category of hazards that must be identified, evaluated, and controlled through the risk management process. The BEP aligns with the risk management plan in defining the scope and approach; the BER outputs (residual biological risks) integrate into the overall residual risk evaluation under ISO 14971 Clause 8.
✦ PREMIUM BUNDLE · EU MDR COMPLETE
The complete EU MDR documentation set — technical, clinical and risk.
The Technical Documentation Kit (Annex II), the Clinical Documentation Kit (Articles 32, 61, 83-86) and the Risk Management Kit (ISO 14971 + IEC 62366-1) in one premium bundle. 22 templates, cross-referenced, with a single Master Index.
- ✓22 templates · pre-market + post-market documentation
- ✓Save €178 vs buying the three kits separately
Conclusions
Biocompatibility testing is one of the most technically demanding areas of medical device compliance — not because the tests themselves are inherently complex, but because the framework requires a genuinely integrated, risk-based approach that connects material science, toxicology, chemistry, and clinical context into a coherent biological evaluation.
The 2025 edition of ISO 10993-1 reinforces this integrated approach — moving definitively away from a checklist mentality toward a scientifically justified, risk-proportionate evaluation that starts with characterization and proceeds to testing only where the data demands it. Manufacturers who invest in strong chemical characterization early in development consistently find that their testing programs are more targeted, their regulatory submissions are better supported, and their overall time to market is faster.
For manufacturers building or upgrading their biological evaluation documentation, the essential starting point is a well-structured BEP and a systematic chemical characterization program. The ISO 10993 Biocompatibility Documentation Package available on MD Regulatory includes a Biological Evaluation Plan template, BER template, device categorization worksheet, and chemical characterization summary template — all aligned with ISO 10993-1:2025 requirements and the EU MDR technical documentation framework.
This article is part of the MD Regulatory series on medical device compliance. Related articles: ISO 10993-1:2025 — What’s New · ISO 14971 Risk Management · EU MDR Technical Documentation · ISO 13485 Complete Guide · Benefit-Risk Analysis