MDSAP Audits: No More Secrets for your Certification
A practical, regulator-aligned walkthrough of the Medical Device Single Audit Program — what it covers, how the audit works, how non-conformities are graded, what it costs in time and effort, and exactly how to prepare.
Quick Answer: What Is an MDSAP Audit?
The Medical Device Single Audit Program (MDSAP) is an international regulatory framework that allows a single audit of a medical device manufacturer’s quality management system (QMS) to satisfy the requirements of five regulatory authorities: Australia (TGA), Brazil (ANVISA), Canada (Health Canada), Japan (MHLW/PMDA) and the United States (FDA). The audit is performed by an authorized Auditing Organization (AO) against ISO 13485 plus country-specific regulatory requirements. A successful audit results in an MDSAP certificate valid for three years, with annual surveillance audits in between.
Table of Contents
What MDSAP Is and Who It’s For
MDSAP stands for Medical Device Single Audit Program. It is a globally recognized scheme that lets a single QMS audit, performed by an authorized Auditing Organization, satisfy the regulatory requirements of multiple participating jurisdictions. Instead of being audited separately by each authority, a manufacturer is audited once, against a unified set of requirements, and the resulting report is accepted by all participating regulators.
MDSAP is relevant for any medical device manufacturer that:
- Already sells, or plans to sell, devices in Canada (where MDSAP is mandatory for Class II, III and IV devices under the Medical Devices Regulations).
- Wants to reduce the regulatory burden of multiple parallel audits in the U.S., Brazil, Japan or Australia.
- Is preparing for FDA inspections and wants the FDA to accept the MDSAP report in lieu of a routine surveillance inspection.
- Operates a multi-site or contract manufacturing model and needs a single, defensible QMS narrative across jurisdictions.
Under the MDSAP framework, a recognized Auditing Organization (AO) performs a single regulatory audit of a medical device manufacturer, which is accepted by the participating Regulatory Authorities (RAs) as evidence of compliance with their respective requirements.
- Australia’s Therapeutic Goods Administration (TGA)
- Brazilian Health Regulatory Agency (ANVISA)
- Health Canada (HC)
- Japan’s Ministry of Health, Labour and Welfare, and the Japanese Pharmaceuticals and Medical Devices Agency(MHLW/PMDA)
- U.S. Food and Drug Administration (USFDA)
Observers and affiliate members (WHO, EU, UK MHRA, South Korea) participate in the program but do not currently accept MDSAP certificates as a replacement for their own conformity assessment processes.
MDSAP audits are conducted by Auditing Organisation towards manufacturer seeking for MDSAP certification is issued by Auditing Organizations (AOs) that have been thoroughly assessed and authorized by a competent Regulatory Authority (RA) to conduct audits and issue MDSAP certificates following the successful completion of the certification process.
MULTI-MARKET KIT
One audit. Five markets. Ready to submit.
Our MDSAP Documentation Kit covers Brazil ANVISA, Japan PMDA, Canada Health Canada, Australia TGA, and FDA — with country-specific reportability worksheets and application checklists you can use today.
- ✓15 SOPs covering 5 MDSAP markets
- ✓18 templates with country worksheets
- ✓Brazil · Japan · Canada · Australia · USA
Structure of the Audit
The audit lifecycle is similar to a standard ISO 13485 certification cycle: a two-stage initial audit, a three-year certificate, and annual surveillance audits — with a recertification audit at the end of the cycle.
Stage 1 — Readiness Review
A documentation-focused review aimed at confirming that the QMS is mature enough to undergo the full audit. Typical Stage 1 outputs include: identification of documentation gaps, confirmation of audit scope (sites, product families, exclusions), and a preliminary view of risk areas. Stage 1 is usually performed on-site at the main manufacturing facility.
Stage 2 — Full QMS Audit
The complete on-site audit, structured around the seven MDSAP process chapters (see Section 5). Auditors sample objective evidence across all relevant processes and assess conformity against ISO 13485 and country-specific regulatory requirements. Non-conformities found here must be addressed with a documented corrective action plan before the certificate can be issued.
Certificate Issuance and Maintenance
Once corrective actions are accepted, the AO issues the MDSAP certificate, valid for three years. During those three years:
- Surveillance audit in Year 1: covers a sub-set of processes plus all critical areas.
- Surveillance audit in Year 2: covers the remaining processes.
- Recertification audit in Year 3: full re-audit of the entire QMS.
| Audit Type | Frequency | Scope |
| Stage 1 | Once, before Stage 2 | Documentation review, readiness check |
| Stage 2 (Initial) | Once, after Stage 1 | Full QMS audit across all 7 process chapters |
| Surveillance Year 1 | 12 months after certification | Partial QMS audit, all critical processes |
| Surveillance Year 2 | 24 months after certification | Remaining processes + follow-up on prior findings |
| Recertification | Every 3 years | Full QMS re-audit, identical scope to Stage 2 |
| Unannounced | As required (e.g. Health Canada) | Triggered by complaints, recalls, or risk signals |
Exclusion of one or more MDSAP Countries
A manufacturer can choose to include only some of the five jurisdictions in the audit scope. This is a strategic decision and must be agreed with the Auditing Organization before the audit plan is finalized.
Common reasons to exclude a country:
- No commercial presence in that market and no plan to enter in the next 3 years.
- The device class is out of scope of the local regulator’s MDSAP recognition (for example, certain IVDs in some jurisdictions).
- A separate, recently passed local inspection is still valid and not worth duplicating.
Important: exclusions reduce audit days and cost, but the resulting certificate will be valid only for the included countries. Adding a country later requires a scope extension audit, not a free amendment.
The structure of the MDSAP Regulation
MDSAP audits follow a documented process model. Every audit, regardless of manufacturer or AO, walks through the same seven chapters. Understanding this sequence is the single most useful thing a quality team can do before the audit.
| Ch. | Process | What Auditors Look For |
| 1 | Management | Management review records, quality policy, resource allocation, regulatory reporting decisions. |
| 2 | Device Marketing Authorization & Facility Registration | Country-specific registrations, UDI, listings, licence renewals, change notifications. |
| 3 | Measurement, Analysis & Improvement | Internal audits, CAPA, complaint handling, data analysis, post-market surveillance feedback loops. |
| 4 | Medical Device Adverse Events & Advisory Notice Reporting | Vigilance procedures per country, MDR/MIR submissions, recall and field safety corrective action records. |
| 5 | Design & Development | Design controls per 21 CFR 820.30, design history file, design changes, verification and validation. |
| 6 | Production & Service Provision | Process validation, environmental controls, sterilization, traceability, servicing records. |
| 7 | Purchasing | Supplier qualification, supplier monitoring, supplier audits, purchasing controls. |
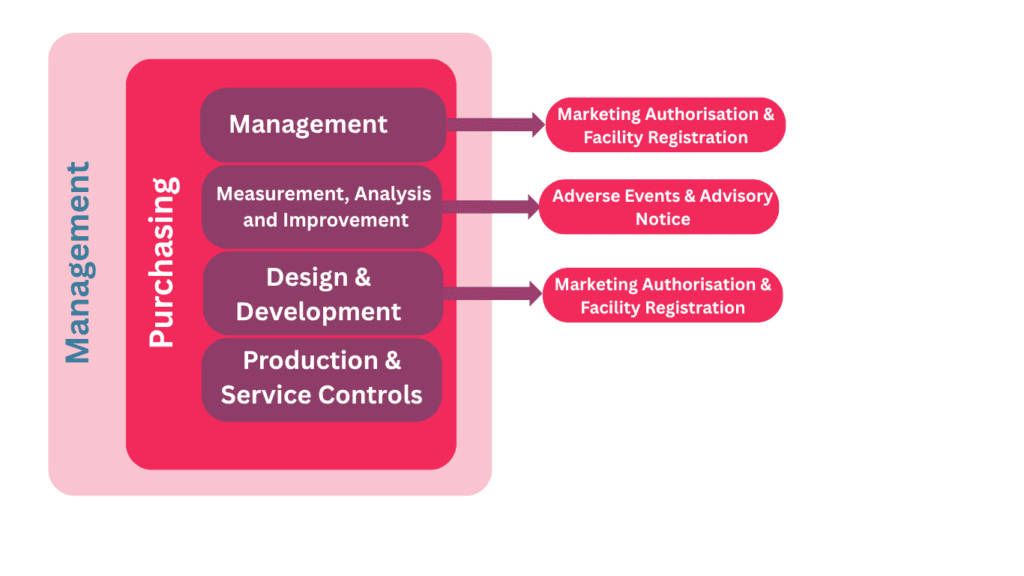
MDSAP process chapters and their interactions
MDSAP vs ISO 13485: Key Differences
MDSAP is built on ISO 13485, but it is not equivalent to it. The differences are where audit teams typically find their hardest findings.
| Dimension | ISO 13485:2016 | MDSAP |
| Scope | QMS for medical devices, jurisdiction-neutral | QMS + jurisdiction-specific regulatory requirements (5 countries) |
| Marketing authorization | Not in scope | Explicitly audited per country (licences, registrations, UDI) |
| Vigilance / adverse events | Generic awareness | Country-specific timelines and submission procedures audited |
| Non-conformity grading | Major / minor (subjective) | Points-based 1–5 scale with documented escalation rules |
| Audit duration | Defined by certification body | Calculated by MDSAP audit time formula (employees, complexity, sites) |
| Unannounced audits | Not required | Possible (especially for Health Canada-driven triggers) |
MDSAP Non-Conformities Grading System
One of MDSAP’s signature features is its objective, points-based grading mechanism for non-conformities. It replaces subjective labels (“significant”, “regular”) with a transparent matrix that produces a numeric grade between 1 and 5.
How the Grade Is Calculated
- Start with the requirement: does the non-conformity affect a clause with direct or indirect impact on the QMS outcome?
- Determine recurrence: is it the first occurrence, or a repeat finding?
- Read the initial grade from the matrix (range 1–4).
- Apply escalation rules: +1 if there is no documented procedure; +1 if non-conforming product has been released.
- Cap the final grade at 5 for reporting purposes.
| Grade | Meaning | Typical Example | Likely Regulator Reaction |
| 1 | Minor, isolated, no QMS impact | Single training record missing for a non-critical role | Logged; no follow-up usually required |
| 2 | Minor with limited QMS impact | Inconsistent records in non-critical supplier evaluation | Tracked at next surveillance audit |
| 3 | Direct QMS impact, first occurrence | Design change not formally controlled in DHF | Corrective action plan required; close-out tracked |
| 4 | Direct QMS impact, recurrent OR escalation | Repeat CAPA closure failure across audits | Heightened regulator attention; possible site action |
| 5 | Severe systemic failure or product safety risk | Release of non-conforming sterile devices without lot review | May trigger certificate suspension, regulator notification |
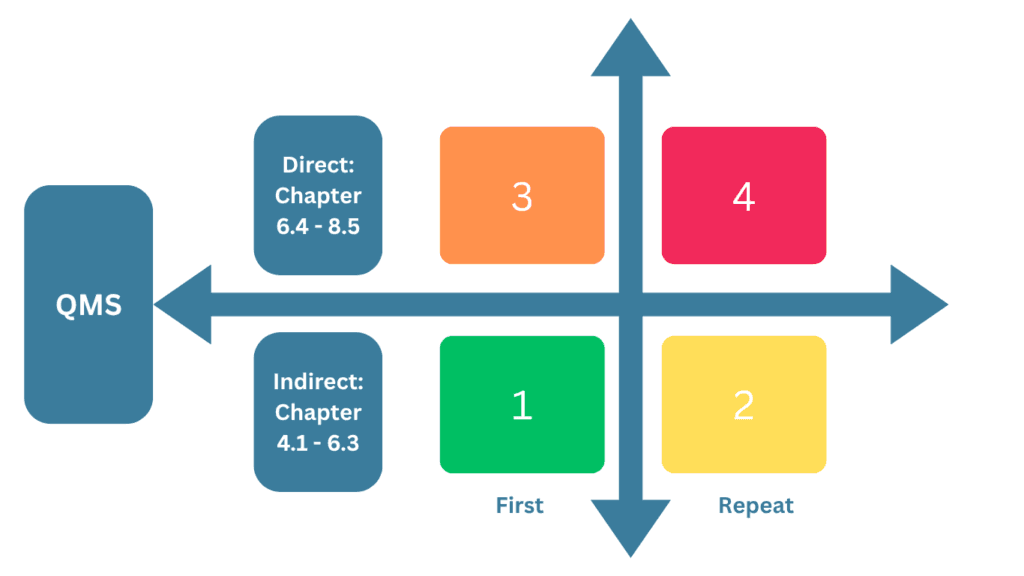
MDSAP non-conformity grading matrix and escalation rules
Market Access Benefits by Country
MDSAP certification plays a key role in facilitating market access for medical device manufacturers by allowing a single, comprehensive audit to satisfy the quality management system requirements of multiple participating regulatory authorities. This approach significantly reduces duplication of audits, optimizes internal resources, and creates a more efficient pathway to global compliance. In certain jurisdictions, such as Canada, MDSAP certification is mandatory for obtaining and maintaining a medical device license, while in others it is widely accepted as a robust demonstration of regulatory compliance, often leading to fewer routine inspections and more streamlined interactions with authorities.
In the context of the United States, although single audit certification is not a formal requirement for market entry, it has a clear and practical connection with the FDA 510(k) process. Companies that maintain an MDSAP-compliant quality management system aligned with 21 CFR Part 820 are typically better prepared to support their 510(k) submissions, as many of the underlying requirements—such as design controls, risk management, and documentation practices—are already addressed through the single audit framework. Furthermore, the U.S. Food and Drug Administration recognizes MDSAP audit reports as a substitute for routine surveillance inspections, which can reduce regulatory burden post-clearance and allow manufacturers to focus on maintaining compliance rather than preparing for separate audits.
The business case for MDSAP varies sharply by market. The matrix below is the practical question a manufacturer should answer before committing.
| Country | Mandatory? | What MDSAP Replaces or Enables |
| Canada | Yes | Required to obtain and maintain a Medical Device Licence (Class II–IV). No MDSAP, no Canadian sales. |
| United States | No | FDA accepts MDSAP reports in lieu of routine surveillance inspections. Strongly aligned with 21 CFR Part 820. |
| Brazil | No (recommended) | Replaces ANVISA’s B-GMP inspection for several device categories; can dramatically shorten time to registration. |
| Japan | No | Used in MHLW/PMDA QMS conformity assessment; reduces overlap with domestic audits. |
| Australia | No | Accepted for TGA conformity assessment for select certification routes. |
Risk-based Approach for MDSAP audits
A risk-based approach to auditing quality systems is closely aligned with the philosophy and structure of the Medical Device Single Audit Program (MDSAP), where audit activities are explicitly designed to focus on processes that have the greatest impact on product quality and patient safety. Rather than applying equal attention to all elements of the quality management system, this approach prioritizes critical areas—such as design controls, production processes, and supplier management—based on their associated risks and their influence on regulatory compliance. This principle is embedded within the MDSAP audit model, which follows a process-based sequence and requires auditors to evaluate not only whether procedures exist, but whether they are effectively implemented in high-risk areas.
A key point of integration is the MDSAP nonconformity grading system, which further reinforces risk-based thinking by assigning severity levels to findings according to their potential impact and systemic nature. This ensures that issues affecting critical processes or indicating broader quality system failures are escalated appropriately, allowing both regulators and manufacturers to prioritize corrective actions. Additionally, these multi-jurisdictional audit program leverages inputs such as complaint data, post-market surveillance, and previous audit results to dynamically adjust audit focus, reflecting the same continuous, data-driven prioritization that defines risk-based auditing.
Ultimately, the connection between risk-based auditing and MDSAP lies in their shared objective: moving beyond checklist compliance toward a deeper, evidence-based evaluation of how well a quality system controls real-world risks. By embedding risk prioritization into both audit execution and nonconformity assessment, MDSAP ensures that audits deliver meaningful insights, support regulatory confidence, and drive continuous improvement in the safety and performance of medical devices.
✦ PREMIUM BUNDLE
The ultimate global QMS documentation bundle.
Combine ISO 13485 + all 5 MDSAP markets in one premium package. Deduplicated structure means you customize each document once — not twice. Save €199 vs buying the kits separately.
- ✓41 SOPs covering both ISO 13485 and MDSAP
- ✓70+ templates with deduplicated structure
- ✓Save €199 vs buying separately
MDSAP Audit Costs and Timelines
Audit duration is calculated using the MDSAP Audit Time formula, which takes into account number of employees, device complexity, number of sites, technology, and outsourced processes. The output is a number of audit days, which the AO then quotes against.
Typical Cost Ranges (Indicative)
| Manufacturer Size | Initial Audit Days | Indicative Initial Cost (Stage 1 + Stage 2) |
| Small (1–25 employees, single site) | 5–7 days | €12,000 – €20,000 |
| Medium (26–100 employees, single site) | 8–12 days | €20,000 – €35,000 |
| Large (100+ employees, multi-site) | 13–20+ days | €40,000 – €80,000+ |
Note: figures are indicative and exclude internal preparation cost, travel, surveillance audits, and remediation. The recurring annual cost (surveillance) is roughly 30–40% of the initial audit cost.
Typical Project Timeline
| Phase | Duration | Key Activities |
| Preparation | 3–9 months | Gap analysis, CAPA, internal audit, mock audit, training |
| AO selection & contract | 1–2 months | Quotation, scope definition, Stage 1 booking |
| Stage 1 | 1–2 days on-site | Documentation review, gap close-out plan |
| Gap remediation | 1–3 months | Address Stage 1 findings before Stage 2 |
| Stage 2 | 5–15 days on-site | Full QMS audit |
| Certification | 1–2 months | Corrective actions, AO review, certificate issuance |
MDSAP Audit Checklist (Excel, Free Download)
The most efficient way to prepare is to work through a MDSAP audit checklist built on the same structure the Auditing Organization will use. This free Excel workbook mirrors the MDSAP Audit Approach (AU P0002): all 90 audit tasks across the seven process chapters — Management, Device Marketing Authorization & Facility Registration, Measurement, Analysis & Improvement, Adverse Events & Advisory Notices, Design & Development, Production & Service Controls, and Purchasing.
Each task lists the official task title, the audit objective, the relevant ISO 13485:2016 clause, and the jurisdiction-specific requirements for Australia (TGA), Brazil (ANVISA), Canada (Health Canada), Japan (MHLW/PMDA) and the USA (FDA QMSR). Use it as a MDSAP requirements checklist and gap analysis tool: record your evidence, set each task to Conform / Nonconformity / Observation / Not Applicable, and the summary sheet counts your findings automatically — including MDSAP nonconformity grades 1–5.
✦ FREE DOWNLOAD · EXCEL
MDSAP Audit Checklist — 90 tasks, 7 process chapters
Built on the MDSAP Audit Approach (AU P0002), with ISO 13485:2016 clauses and requirements for all five jurisdictions (TGA, ANVISA, Health Canada, MHLW/PMDA, FDA QMSR). Editable Excel, with automatic findings summary and grades 1–5.
↓ Download the checklist (Excel)Need the full document set behind these tasks? Our MDSAP Documentation Kit provides the SOPs and templates — CAPA, internal audit, management review, adverse-event reportability worksheets and country-specific application checklists — mapped to the same audit model.
Preparation Strategy for MDSAP
MDSAP preparation is not a documentation sprint; it is a 6–12 month operational alignment. The checklist below mirrors how AOs structure their audits, so working through it in order maximizes return on effort.
Months -12 to -9: Foundation
- Gap analysis against the MDSAP Companion Document and each of the five jurisdictional requirements.
- Map your QMS to the seven MDSAP process chapters — most ISO 13485 QMS files are organized by clause, not by process, and this remapping is often underestimated.
- Select the AO early; lead times can exceed 6 months for Stage 1 booking with major AOs (BSI, DEKRA, TÜV SÜD, DNV, Intertek, etc.).
Months -9 to -6: Country-Specific Alignment
- Document jurisdiction-specific vigilance procedures (FDA MDR, Health Canada MPR, ANVISA, TGA, PMDA — each with their own timelines).
- Verify all marketing authorizations are current: U.S. registration & listing, Canadian MDL, ANVISA registration, J-MDN code, TGA ARTG entry.
- Map UDI assignments and country-specific labelling requirements.
Months -6 to -3: Internal Validation
- Conduct a full internal audit using the MDSAP process sequence — not the old ISO 13485 clause-based audit.
- Run a mock audit, preferably with an external consultant familiar with MDSAP grading.
- Close all open CAPAs older than 6 months. Open CAPAs are the #1 source of grade-3 findings.
Months -3 to 0: Final Readiness
Prepare the data room: complaint logs, CAPA list, supplier evaluation status, design changes, post-market surveillance summaries.
Train front-line staff on what to say (and not say) during interviews — especially production operators and complaint handlers.
Common Audit Findings and How to Avoid Them
Across published MDSAP audit summaries, the same families of findings recur year after year. Pre-emptive remediation of these areas measurably reduces audit risk.
| Finding Area | Typical Issue | Quick Mitigation |
| CAPA effectiveness | Closed CAPAs reopen because root cause was symptomatic, not systemic | Require independent reviewer to sign off on effectiveness check before closure |
| Design changes | Changes implemented in production without DHF update | Lock the change control workflow to a DHF gate |
| Supplier management | Critical suppliers re-evaluated only once, at qualification | Annual supplier review with quantitative criteria |
| Vigilance reporting | Domestic reporting OK, but missing country-specific MDR/MIR submissions | Country-by-country decision tree maintained alongside complaint procedure |
| Marketing authorization | Licence amendments lag behind product changes | Trigger licence review at every Class B and above design change |
Future Perspective
The future of the Medical Device Single Audit Program (MDSAP) is expected to be shaped by a continued evolution toward greater regulatory convergence and increased reliance by participating authorities on audit outcomes as a primary oversight tool. As global regulatory expectations become more aligned, MDSAP requirements are likely to expand in depth and scope, with stronger emphasis on areas such as post-market surveillance, real-world evidence, and data integrity, reflecting broader trends in medical device regulation. At the same time, regulatory bodies are progressively leveraging MDSAP audit reports to reduce the need for routine inspections, signaling a shift toward a more efficient, audit-driven compliance model. This growing reliance means that the quality and consistency of MDSAP audits—and the robustness of manufacturers’ quality systems—will play an increasingly critical role in maintaining market access. Looking ahead, organizations that invest early in strengthening their processes, digitalizing quality systems, and adopting a risk-based mindset will be better positioned to adapt to future updates and benefit from a regulatory environment that is moving toward greater harmonization and trust in unified audit frameworks.
Frequently Asked Questions
Is MDSAP mandatory?
Only in Canada, where it is required for Class II, III and IV medical device licences. In the other four jurisdictions it is voluntary but actively recognized.
How long does an MDSAP certificate last?
Three years from the date of issue, with mandatory surveillance audits in Year 1 and Year 2, and a full recertification audit in Year 3.
Can I keep my existing ISO 13485 certificate and add MDSAP?
Yes. Most AOs offer combined audits where the ISO 13485 surveillance and MDSAP surveillance are performed in a single visit, reducing duration and cost. The certificates remain separate but the audit is integrated.
Does MDSAP replace the FDA QSR (21 CFR Part 820)?
No. MDSAP audits include 21 CFR Part 820 in their scope, so a compliant MDSAP QMS is also Part 820-compliant — but the underlying regulation remains in force. With the FDA’s QMSR (Quality Management System Regulation) harmonizing Part 820 with ISO 13485 starting in February 2026, the overlap is becoming even tighter.
What happens if I receive a Grade 4 or 5 non-conformity?
The AO will require a comprehensive corrective action plan, with a tight close-out timeline. The regulator(s) most affected by the finding are notified through the standardized audit exchange form. In severe cases, the certificate can be suspended or withdrawn until remediation is verified.
Which Auditing Organizations are authorized?
The official list is maintained by the MDSAP Regulatory Authority Council. Major authorized AOs include BSI, DEKRA Certification, DNV, Intertek, NSF International, SGS, TÜV Rheinland, TÜV SÜD and UL Solutions. Selection should be based on geographic coverage, sector experience and lead times — not price alone.
How much does an MDSAP audit cost?
Initial certification typically ranges from €12,000 for a very small single-site manufacturer to €80,000+ for large multi-site operations. Recurring surveillance audits cost roughly 30–40% of the initial audit per year. Internal preparation cost is usually equal to or greater than the AO fee.
Conclusions
MDSAP started as an administrative consolidation — five audits collapsed into one. It has matured into something more strategic: a single, defensible narrative of how a manufacturer controls quality across the most demanding regulatory markets in the world.
Manufacturers that treat MDSAP as a compliance overhead will pay for it twice — once in audit fees, and again in remediation. Those that treat it as the operational backbone of their global quality system will find that the same investment unlocks Canada, smooths the FDA, accelerates Brazil and removes friction from Japan and Australia. The audit is not the deliverable. The audit is the proof that the system already works.