IEC 62304 Software Verification and Validation: Requirements by Safety Class
Table of Contents
Introduction
The title of this article reflects how the industry talks, not how the standards define the terms. IEC 62304 software verification validation is the search term most engineers type, but IEC 62304 itself covers verification only — software validation as a regulatory concept lives in a different set of documents. Confusing the two is the most common technical error in software regulatory submissions, and it is the source of countless Notified Body and FDA findings. This guide separates the two cleanly, then walks through what IEC 62304 actually requires for verification at each safety class, what testing rigor is expected in practice, how documentation and traceability must be structured, and where validation responsibilities sit outside IEC 62304.
The article assumes familiarity with the basics of medical device software lifecycle processes; the IEC 62304 complete guide covers the broader framework, and SOUP management under IEC 62304 addresses the parallel obligations on third-party software components. The verification activities described here apply to software developed in-house; SOUP follows a different evidence model.
What IEC 62304 actually covers — and what it does not
IEC 62304 is a process standard for the medical device software lifecycle. It defines what activities must be performed during software development, maintenance and risk management, and — crucially — it defines those activities at three levels of rigor based on software safety class. The verification activities are concentrated in clauses 5.5 (software unit implementation and verification), 5.6 (software integration and integration testing) and 5.7 (software system testing), with traceability obligations across clauses 5.1 to 5.8 and software risk management in clause 7.
What IEC 62304 does not cover, despite frequent assumption to the contrary, is software validation in the regulatory sense. The standard’s foreword and Annex B make this explicit: IEC 62304 addresses the development of medical device software as a component or as a standalone product, but the validation that the device or system as a whole meets user needs and intended use is the responsibility of other standards and processes. Software validation lives in three places: ISO 13485 clause 7.3.7 (design and development validation), IEC 82304-1 (health software product safety, which sits above IEC 62304 for standalone software), and IEC 62366-1 (usability engineering, which validates that the user can operate the software safely and effectively).
This separation is not academic. A common scenario: a manufacturer submits a software design history file in which “verification and validation” are presented as a single unified process under IEC 62304. The Notified Body raises a major non-conformity because the validation evidence — that the software meets user needs and intended use in the actual use environment — is missing or buried inside what is actually verification testing. The fix is to restructure the documentation along the correct standard boundaries: IEC 62304 verification proves that what was built matches the specification; ISO 13485 / IEC 82304-1 / IEC 62366-1 validation proves that the specification was the right one.

Figure 1 — How software verification (IEC 62304) and software validation (ISO 13485, IEC 82304-1, IEC 62366) sit in the medical device software lifecycle.
✦ AI/ML DOCUMENTATION KIT
One AI/ML technical file, five markets — built around the PCCP reviewers call the hardest to draft.
10 coordinated documents covering the full AI lifecycle: development & lifecycle plan, data governance & bias management, model validation, AI risk management, the flagship Predetermined Change Control Plan (PCCP), human oversight & transparency, postmarket performance monitoring, an AI-augmented Clinical Evaluation Plan & Report, and the AI/ML development SOP — with the IMDRF GMLP principles as the convergent spine.
- ✓10 documents · 9 Word + 5-sheet Master Index & Conformity Matrix (Excel)
- ✓GMLP spine across FDA · EU AI Act · Health Canada · TGA · ANVISA
Software safety classes: the gating decision
Every verification activity required by IEC 62304 is gated by software safety class. The standard defines three classes, assigned at the level of the software system as a whole and inherited by software items unless segregation is implemented and documented.
Class A applies when no injury or damage to health is possible from a software failure. Class B applies when injury that is not serious is possible. Class C applies when death or serious injury is possible. The classification is performed before development begins, on the basis of a hazardous situation analysis: the manufacturer identifies the hazardous situations that could arise from software contributing to the device, evaluates the severity of the resulting harm assuming no risk control measures are in place, and assigns the class accordingly. Risk control measures implemented in software cannot reduce the safety class of the software that implements them — a defensive interlock written in software does not downgrade the controlled function. Risk controls outside the software (hardware interlocks, mechanical limits, independent monitoring devices, clinical procedures) can reduce the safety class, provided the segregation is architecturally demonstrated.
The 2015 amendment to IEC 62304 introduced two important nuances. First, it clarified that software components inside a system can carry different safety classes provided the architecture demonstrates effective segregation — a Class A logging utility inside a Class C infusion pump is acceptable if the architectural and design analysis shows the logging cannot affect therapy delivery. Second, it made software system testing mandatory for all classes including Class A. Pre-amendment guidance allowed Class A to omit system testing; post-amendment, every class must perform and document system testing.
The classification decision drives the entire verification effort. Misclassification — particularly under-classification — is a leading cause of regulatory rework: software classified as B but later determined to be C requires retroactive generation of detailed design documentation, unit verification evidence, integration test records and traceability that may not exist. The cost of reconstructing this evidence after the fact, with fragmented memory of design decisions, regularly exceeds the cost of generating it correctly during development. Conservative classification, particularly for software with clinical functions, is almost always the right risk decision.

Figure 2 — IEC 62304 verification requirements by software safety class, showing which clauses apply and the documentation expected at each class.
Software unit verification: clause 5.5
Software unit verification applies to Class B and Class C software. A unit, in IEC 62304 terms, is the smallest software item not subdivided further by the development team — typically a function, method, class or module depending on the language and architecture. The standard does not prescribe what constitutes a unit; the manufacturer defines this in the development plan, and the definition must be consistent with the granularity at which the detailed design is documented.
For Class B, clause 5.5 requires that software units be implemented and verified. The verification activities are not exhaustively prescribed but must be planned and documented. Practical verification at Class B includes static analysis (compiler warnings at high level, lint, code review against coding standards), unit-level testing of functions whose behaviour can be tested in isolation, and code review focused on adherence to architecture and detailed design.
For Class C, clause 5.5.5 adds the requirement that acceptance criteria for unit verification be defined, that the criteria be applied, and that the results be evaluated against the criteria. This is the language Notified Bodies cite when raising findings on Class C software with weak unit-level evidence. Acceptance criteria typically address: code coverage thresholds (statement, branch, modified condition/decision coverage as appropriate to the language and risk); compliance with the coding standard; absence of static analysis findings above a defined severity; and successful execution of unit tests against documented expected results.
The level of testing rigor expected in practice has stabilised over the past decade. For Class C software in safety-critical functions, the de facto industry expectation is statement coverage above 95% and branch coverage above 90%, supported by automated unit test execution in the build pipeline. For Class B, statement coverage above 80% with documented justification for uncovered code is typically defensible. These numbers are not in IEC 62304 — the standard requires acceptance criteria for Class C but does not specify thresholds — but they reflect the rigor that auditors have come to expect.
Tool qualification is a related obligation. Compilers, static analysers, unit test frameworks and coverage tools used to generate verification evidence affect the trustworthiness of that evidence. For Class C, tool qualification should be documented at a level proportionate to the tool’s role: a tool whose output is independently checked (a compiler whose output is system-tested) requires lighter qualification than a tool whose output is the verification evidence itself (a coverage tool whose numbers are the basis for acceptance). A short written tool assessment per tool, recording the manufacturer’s name, version, intended use, qualification rationale and any known limitations, is normally sufficient and is expected.
Software integration and integration testing: clause 5.6
Integration testing applies to Class B and Class C. Its objective is to verify that the integrated software items behave as specified by the architectural design, and that the interfaces between items operate correctly. Integration is performed in stages defined by the integration plan — typically bottom-up from low-level components toward the system level — and integration testing accompanies each integration stage.
The content of integration tests is driven by the architectural design. Each defined interface between software items requires test cases covering nominal behaviour, boundary conditions, and error handling. Where the architecture documents segregation between software items of different safety classes, the integration testing must also demonstrate that the segregation behaves as designed under fault conditions: a failure in a Class A item must be shown not to affect a Class C item if the architecture claims segregation as a risk control.
Integration testing is documentation-heavy in a way that unit verification typically is not. Each test must have a documented procedure, expected result, actual result, and pass/fail evaluation, plus traceability back to the architectural element being tested. Integration test failures must be recorded as anomalies, investigated under the configuration management and problem resolution processes, and re-tested after fix. This evidence trail is one of the first things audited in a software design history file review, and gaps in it are common findings.
Software system testing: clause 5.7
System testing verifies that the software system satisfies its software requirements. Since the 2015 amendment, this activity is mandatory for all classes, including Class A. The testing covers the software running in its target environment — or in a target-equivalent environment with documented justification — and exercises the functional, performance, interface and safety requirements identified in clause 5.2.
The most frequent error in system test design is to test against requirements only, omitting risk-derived acceptance criteria. Clause 7 of IEC 62304 requires that risk control measures implemented in software be verified, and clause 5.7.4 explicitly requires that system testing verifies the implementation of risk control measures. A robust system test plan structures its test cases by source: functional requirements traced to test cases, performance requirements traced to test cases, interface requirements traced to test cases, and risk control measures traced to test cases. The traceability matrix (more on this in the next section) is the artefact that makes this completeness visible.
System testing must produce documented evidence. Each test case should be executed against a controlled software build, the results recorded with sufficient detail to reproduce the test, anomalies logged, and the final test report signed off. For Class C, regression testing must demonstrate that previously verified functionality still works after changes; the regression test suite is itself a controlled artefact and must be maintained alongside the software.
For software released as part of a finished medical device, system testing under IEC 62304 is not equivalent to design validation under ISO 13485 clause 7.3.7. System testing demonstrates that the software meets its specifications. Validation demonstrates that the device — software and all — meets user needs and intended use, in the actual or representative use environment, by the actual or representative user population. Validation requires clinical evaluation, usability validation per IEC 62366-1, and where applicable simulated or actual use testing. The system test report and the validation report are separate artefacts with separate audiences.
Traceability: the spine of the verification record
Traceability is the single artefact that ties the verification effort together and the artefact most likely to be examined first by an auditor. Clause 5.1.1 requires that the manufacturer establishes traceability among the software lifecycle outputs, and the granularity of traceability scales with safety class.
For Class A, traceability links software requirements to system tests. For Class B, traceability extends to architecture: software requirements trace to architectural items, architectural items trace to integration tests, software requirements trace to system tests. For Class C, traceability is full and bidirectional: software requirements ↔ architecture ↔ detailed design ↔ unit tests ↔ integration tests ↔ system tests, with the addition of risk control measures traced through to verification evidence.
The traceability matrix is normally maintained as a living artefact in a tool — application lifecycle management platforms, requirements management tools, or for smaller projects controlled spreadsheets — and updated continuously as requirements, design and tests change. A common Notified Body finding is a traceability matrix produced as a one-time deliverable at the end of development, inconsistent with the actual artefacts in the design history file. The matrix must be updated under change control and included in the release evidence.
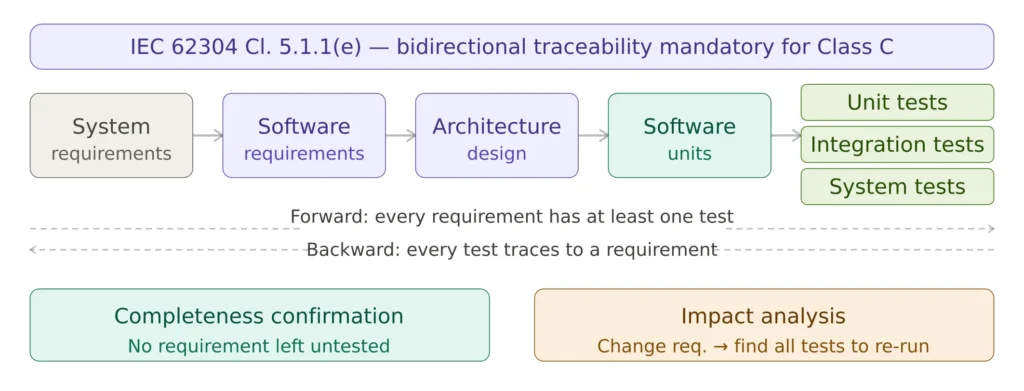
Figure 3 — Bidirectional traceability matrix structure for IEC 62304 Class C software, linking requirements through design and verification.
Risk control verification under clause 7
Software risk management runs in parallel to development under clause 7 of IEC 62304, integrated with the device-level ISO 14971 risk management process. For verification purposes, two clause 7 obligations matter most.
First, every risk control measure implemented in software must be verified. The verification is normally embedded in the system testing or integration testing rather than performed as a separate activity, but the test cases must be explicitly identified as covering the risk control measure, and the traceability from risk control measure to test case must exist. A risk control measure for which no specific verification evidence can be produced is a finding, regardless of how robust the rest of the verification is.
Second, where the architecture claims segregation between software items as a risk control — for example, segregating a Class A logging utility from a Class C therapy delivery item to prevent the logging from affecting therapy — the segregation itself must be verified. Architectural verification of segregation typically includes static analysis evidence that the segregated items do not share state inappropriately, dynamic test evidence that fault injection in one item does not propagate to the other, and review evidence that the runtime environment (operating system isolation, memory protection, scheduling priority) supports the claimed segregation. Segregation claimed but not verified will not be accepted as a basis for downgrading software safety class.
✦ EU MDR TECHNICAL DOCUMENTATION KIT
The complete Annex II technical file in one coordinated set — built to Notified Body review expectations.
8 professionally drafted Word templates covering the full structure of EU MDR Annex II: Device Description, Labelling & IFU, Design & Manufacturing Information, General Safety & Performance Requirements, Benefit-Risk & Risk Management, Verification & Validation, Post-Market Surveillance (Annex III) and the Declaration of Conformity (Annex IV) — with Team-NB position-paper requirements integrated as inline notes.
- ✓8 Word templates · Master Index (Excel) + README
- ✓Full Annex II + Annex III (PMS) + Annex IV (DoC) · MDCG & Team-NB aligned
Documentation deliverables
The verification effort produces a set of controlled documents that together form the verification evidence of the software design history file. The required deliverables scale by class.
The software development plan defines what verification activities will be performed, by whom, with what tools, against what criteria, and how evidence will be captured. The plan is an output of clause 5.1 and is mandatory for all classes; for Class B and C it must address each of the verification clauses applicable to that class.
The software requirements specification is the input against which system testing is performed. Requirements must be uniquely identified, individually testable, and traceable. Vague or untestable requirements are a frequent root cause of weak verification evidence — a test cannot produce conclusive evidence against a requirement that does not specify what “correct” means.
The architectural design specification (Class B and C) and the detailed design specification (Class C) are the inputs against which integration and unit verification respectively are performed. These documents must define the software items and their interfaces with sufficient precision that test cases can be designed against them.
The verification plan and procedures document the planned verification, typically structured by clause: unit verification plan, integration test plan, system test plan. Each plan defines the scope, approach, environment, acceptance criteria, and reporting format.
The verification reports record the executed verification: tests run, evidence captured, anomalies identified, evaluation against acceptance criteria, and the verifier’s signed conclusion. Reports must reference the controlled software build under test, the controlled test procedure, and the controlled environment.
The traceability matrix is the cross-reference between requirements, design, risk controls and verification evidence, maintained continuously throughout development.
The software release record under clause 5.8 closes the verification effort: it confirms that all planned verification activities have been completed, that all anomalies have been resolved or accepted with documented rationale, and that the release authorisation is granted by the responsible signatories.
FDA documentation levels and the QMSR convergence
FDA’s expectations on software documentation have historically been organised around documentation levels — formerly “levels of concern”, now “Basic” and “Enhanced” documentation under the September 2023 FDA guidance “Content of Premarket Submissions for Device Software Functions”. The documentation levels are not the same as IEC 62304 safety classes, but the underlying principle is identical: higher software risk requires more comprehensive documentation.
A practical mapping is that Class A software typically aligns with FDA Basic documentation, and Class B / Class C software typically requires Enhanced documentation, but the alignment is not deterministic. The FDA expects manufacturers to assess the documentation level on the basis of the device’s intended use, the contribution of the software to potential harm, and the regulatory submission pathway. Software classified as Class B under IEC 62304 may still require Enhanced documentation if the software function has a direct role in clinical decision-making or therapy delivery.
With FDA’s adoption of the Quality Management System Regulation effective 2 February 2026, ISO 13485:2016 is incorporated by reference into US medical device QMS requirements. IEC 62304 is referenced as a recognized consensus standard under FDA’s standards recognition process. Authoritative reading on FDA’s recognized standards database is available via the FDA recognized standards portal. The practical effect for manufacturers serving both EU and US markets is that a verification evidence package built to IEC 62304 expectations, with traceability and documentation level appropriate to the risk, will substantially satisfy both regulators.
This is the bridge to the cybersecurity cluster: software verification under IEC 62304 establishes that the software meets its functional and performance requirements, but it does not establish that the software is secure against cyber threats. Cybersecurity testing and risk management is addressed by IEC 81001-5-1 and is a complementary obligation, not a substitute. A Class C software with strong functional verification but weak cybersecurity testing fails the regulatory bar at both Notified Body and FDA review. For the testing methodologies, see medical device cybersecurity testing.
Common audit findings on software verification
Across software design history file reviews and Notified Body audits, a small set of findings recurs with frequency that reveals systemic gaps in how manufacturers approach IEC 62304 verification. Each has a known prevention.

Figure 4 — The six most common IEC 62304 verification audit findings, with the practical fix for each.
Frequently asked questions
What is the difference between software verification and software validation under IEC 62304?
IEC 62304 covers verification only — the activities that demonstrate the software has been built according to its specifications, including unit verification (5.5), integration testing (5.6), and system testing (5.7). Software validation in the regulatory sense — demonstrating that the software meets user needs and intended use in the actual use environment — sits in different standards: ISO 13485 clause 7.3.7 (design validation), IEC 82304-1 (health software product safety), and IEC 62366-1 (usability engineering). Conflating the two is the most common documentation error in software regulatory submissions.
Does IEC 62304 require unit testing for Class A software?
No. Unit verification under clause 5.5 applies to Class B and Class C only. For Class A, the standard requires only software requirements, software release, and — since the 2015 amendment — software system testing. Architecture, detailed design, unit verification and integration testing are not mandatory at Class A. Manufacturers may still perform these activities as good engineering practice, but they are not regulatory requirements at Class A.
What code coverage is required for Class C software under IEC 62304?
The standard does not specify code coverage thresholds. Clause 5.5.5 requires acceptance criteria for unit verification at Class C, with the criteria themselves defined by the manufacturer in the development plan. Industry practice for Class C in safety-critical functions has stabilised around statement coverage above 95% and branch coverage above 90%, with stricter coverage (modified condition/decision) for highest-risk components. The chosen thresholds must be documented, justified against the software risk, and consistently applied — auditors check that what the plan says is what the records show.
Is system testing mandatory for Class A software?
Yes. The 2015 amendment to IEC 62304 made software system testing under clause 5.7 mandatory for all classes including Class A. Pre-amendment guidance allowed Class A to omit system testing; this is no longer correct. A frequent audit finding is Class A software with no documented system test report — this is a major non-conformity post-2015.
How does IEC 62304 verification align with FDA documentation levels?
FDA documentation levels (Basic and Enhanced under the 2023 software submission guidance) are not the same as IEC 62304 safety classes, but the principle of risk-proportionate documentation is shared. A typical mapping is Class A to Basic and Class B / Class C to Enhanced documentation, but the alignment is not deterministic — FDA may require Enhanced documentation for Class B software with a direct clinical role. The QMSR adoption effective 2 February 2026 incorporates ISO 13485:2016 by reference and recognises IEC 62304 as a consensus standard, so a verification package built to IEC 62304 with risk-appropriate documentation will substantially satisfy both EU and US regulators.
Can I use the same evidence for IEC 62304 verification and ISO 13485 design validation?
Generally no. Verification evidence under IEC 62304 demonstrates conformity to specifications. Validation evidence under ISO 13485 7.3.7 demonstrates that the device meets user needs and intended use in the actual or representative use environment. The audiences and acceptance criteria are different. Some artefacts may overlap — a system test executed in a clinically representative environment with users from the target population can contribute evidence to both — but the reports must address the two purposes separately, with separate sections, separate acceptance criteria and separate sign-offs.
What traceability is required for Class B software?
For Class B, traceability under clause 5.1.1 connects software requirements to architectural items and to system tests, and architectural items to integration tests. Detailed design and unit-level traceability are not required at Class B. The matrix should be maintained under change control as a living artefact and demonstrate completeness in both directions: every requirement traces to at least one test, and every test traces to at least one requirement.
Do I need to qualify the tools used for software verification?
Yes, for Class B and especially Class C. Compilers, static analysers, unit test frameworks and coverage tools used to generate verification evidence affect the trustworthiness of that evidence. The qualification level is proportionate to the tool’s role — tools whose output is independently checked require lighter qualification than tools whose output is the verification evidence itself. A short documented tool assessment per tool, recording the manufacturer, version, intended use, qualification rationale and known limitations, is normally sufficient and is expected at audit.
Conclusions
A defensible IEC 62304 verification effort begins with three correct decisions made early. First, the software is correctly classified, with conservative judgement where the contribution to potential harm is uncertain. Second, the verification activities are scoped to the class — not over-engineered for low-risk software, not under-engineered for Class C — and documented in the development plan with explicit acceptance criteria. Third, the boundary between IEC 62304 verification and ISO 13485 / IEC 82304-1 / IEC 62366 validation is held throughout the documentation, so that auditors find the verification evidence where they expect it and the validation evidence where they expect it.
Around these three decisions, the operational discipline is consistent: traceability maintained as a living artefact in an ALM tool, system testing for all classes, acceptance criteria for Class C unit verification, explicit verification of risk control measures, tool qualification documented for verification-supporting tools, and a release record under clause 5.8 that closes every loop opened during development. With FDA QMSR adoption effective 2 February 2026, this discipline serves both EU MDR and US regulatory pathways with a single coherent evidence package, and feeds directly into the cybersecurity obligations under IEC 81001-5-1 that complete the modern medical device software regulatory profile.
The IEC 62304 Software Lifecycle Documentation Package on MD Regulatory includes the software development plan template, software requirements specification template, architecture and detailed design templates, unit verification and integration test plan templates, system test plan and report templates, traceability matrix template, software risk management plan, and software release record — all aligned with IEC 62304:2006/A1:2015, IEC 82304-1, FDA QMSR and EU MDR requirements and immediately deployable in an existing ISO 13485 quality management system.
For the broader software lifecycle context, see the IEC 62304 complete guide. For the parallel obligations on third-party software components, see SOUP management under IEC 62304. For the cybersecurity obligations that sit alongside functional verification, see IEC 81001-5-1 cybersecurity for health software.