EU MDR Technical Documentation Requirements: Complete Guide to Annex II and III
EU MDR technical documentation requirements define the full scope of evidence that every medical device manufacturer must compile, maintain, and make available to demonstrate compliance with Regulation (EU) 2017/745. Unlike the old Medical Device Directive, which allowed relatively lean technical files, the MDR demands a substantially more comprehensive and rigorous body of documentation — one that must tell a coherent, traceable story connecting device design, clinical evidence, risk management, and post-market surveillance into a single integrated system.
This article provides the most complete guide available to EU MDR technical documentation under Annex II and Annex III: what each section requires, how the requirements differ from the MDD, what Notified Bodies are finding in practice, and how to build a technical documentation system that is audit-ready and commercially defensible.
What Is EU MDR Technical Documentation?
The EU MDR technical documentation — previously referred to as the “technical file” under the MDD — is the complete body of evidence that a manufacturer must compile to demonstrate that a medical device conforms with all applicable requirements of EU MDR 2017/745. It is the central artifact of the EU conformity assessment process and the primary document that Notified Bodies review when issuing CE certificates.
EU MDR technical documentation is structured across two annexes:
- Annex II covers the main body of technical documentation — device description, design and manufacturing information, general safety and performance requirements (GSPRs) compliance, benefit-risk analysis and risk management, product verification and validation, and clinical evaluation
- Annex III covers the post-market surveillance technical documentation — specifically the PMS plan, Periodic Safety Update Report (PSUR) or Post-Market Surveillance Report (PMSR), and the Summary of Safety and Clinical Performance (SSCP) for implantable and Class III devices
Together, Annexes II and III define everything a manufacturer needs to demonstrate MDR compliance. Every manufacturer — regardless of device class — must compile technical documentation meeting these requirements. Even Class I non-sterile, non-measuring, non-reusable devices that do not require Notified Body involvement still need technical documentation, as competent authorities may ask to see it at any time.
Who Needs EU MDR Technical Documentation?
Technical documentation under Annexes II and III is required for every medical device placed on the EU market, without exception. The key differences are in who reviews it and how:
Class I devices (non-sterile, non-measuring, non-reusable): Self-certified by the manufacturer. No Notified Body involvement in the technical documentation review, but documentation must still fully comply with Annex II and III requirements and must be available for competent authority inspection.
Class Is, Im, Ir (sterile, measuring, reusable surgical instruments) and Class IIa, IIb, III: Notified Body involvement is required. The Notified Body reviews technical documentation as part of the conformity assessment process — either comprehensively or on a sampling basis, depending on device class and conformity assessment route.
Class III and implantable devices: The most rigorous review. For Class IIb implantable devices, technical documentation review is performed on 100% of devices rather than a representative sample. Class III devices may also require consultation with expert panels for novel technologies or devices without an established clinical history.

Figure 1 — EU MDR technical documentation structure: Annex II and Annex III
Annex II — Section by Section Breakdown
Section 1 — Device Description and Specification
MDR Annex II Section 1.1 — Device Description and Specification (Sub-elements a–j)
Notified Body reviewers frequently check the technical file against the exact sub-lettering of Annex II Section 1.1. A compliant Section 1.1 must address each of the following, in order:
- 1.1(a) — product or trade name and a general description of the device, including its intended purpose and intended users;
- 1.1(b) — the Basic UDI-DI assigned by the manufacturer, or a clear identification by means of product code, catalogue number or other unambiguous reference allowing traceability;
- 1.1(c) — the intended patient population and the medical conditions to be diagnosed, treated and/or monitored, together with patient selection criteria where appropriate, indications, contra-indications and warnings;
- 1.1(d) — the principles of operation of the device and its mode of action, scientifically demonstrated where necessary;
- 1.1(e) — the rationale for the qualification of the product as a device;
- 1.1(f) — the risk class of the device and the justification for the classification rule(s) applied in accordance with Annex VIII;
- 1.1(g) — an explanation of any novel features;
- 1.1(h) — a description of the accessories, other devices and other products that are not devices, which are intended to be used in combination with it;
- 1.1(i) — a description or complete list of the various configurations/variants of the device intended to be made available on the market;
- 1.1(j) — a general description of the key functional elements (parts/components, including software where relevant), its formulation, composition and functionality, with labelled pictorial representations (diagrams, photographs, drawings) where relevant.
Section 1.2 then requires a reference to previous and similar device generations: an overview of the manufacturer’s previous generation(s) of the device where these exist, and an overview of identified similar devices available on the Union or international markets.
This is the foundation of the entire technical documentation. It must provide a complete and unambiguous description of the device, including:
Device name and type: The trade name, common name per EMDN (European Medical Device Nomenclature), and the device family or range covered by the documentation.
Intended purpose: One of the most scrutinized elements by Notified Bodies. The intended purpose must be precisely defined — specifying the medical condition being treated, diagnosed, or monitored; the patient population (age groups, disease stages, contraindications); the body parts or tissue types the device interacts with; and the clinical benefits the device is intended to provide. Unclear device intended use is one of the most common issues cited in Notified Body submissions, contributing to extended review lead times.
Device variants and accessories: Every variant (different sizes, configurations, materials) and every accessory that forms part of the device system must be documented and identified with its specific UDI-DI.
Device description and principle of operation: A technical explanation of how the device works — its mechanism of action, physical and technical characteristics, materials in contact with the patient, software components (if any), and images or diagrams illustrating the device.
Novelty: For novel devices or novel clinical applications, explicit documentation of what is new and why it introduces specific risks that must be addressed.
UDI: The Unique Device Identifier assigned to the device and each variant, with the corresponding EUDAMED registration.
Section 2 — Design and Manufacturing Information
This section establishes the traceability chain from design specifications through to manufactured product. It must include:
Design stages: Documentation of the complete design and development process, aligned with the design controls requirements of ISO 13485 and MDR Article 10. This includes design inputs, design outputs, design review records, design verification and design validation evidence.
Manufacturing information: The name and address of all manufacturing sites, a description of the manufacturing methods and processes for critical components, and evidence of manufacturing process validation where applicable.
Sterilization: For sterile devices, the sterilization method, validation data, and sterility assurance level (SAL) documentation.
Traceability: A complete supplier list for critical components and materials, with evidence of supplier qualification and control.
Section 3 — General Safety and Performance Requirements (GSPRs)
The GSPRs are defined in Annex I of EU MDR 2017/745 and cover 23 numbered requirements across three chapters: general requirements, requirements regarding design and manufacture, and requirements regarding information supplied with the device.
Demonstrating GSPR compliance typically takes one of two forms:
Conformity with harmonized standards: If the manufacturer applies harmonized European standards (EN ISO standards published in the Official Journal of the EU), they benefit from a presumption of conformity with the corresponding GSPR. The technical documentation must list every standard applied, the version used, and — critically — any deviations from the standard with justification.
Common specifications (CS): Where harmonized standards do not exist or are insufficient, the European Commission may publish common specifications that are legally binding.
Other means: Where neither harmonized standards nor common specifications exist, the manufacturer must demonstrate compliance through other appropriate means — literature, testing, or expert opinion — with full documentation and justification.
The GSPR checklist in the technical documentation must cover every applicable GSPR, explicitly demonstrating how each requirement is met, referencing the specific evidence (test reports, standards, literature) that supports the claim of compliance.
Section 4 — Benefit-Risk Analysis and Risk Management
Risk management under EU MDR must follow ISO 14971:2019 throughout the entire product lifecycle. The technical documentation must include:
Risk management plan: Defining the scope, responsibilities, and risk acceptability criteria for the device.
Risk analysis: Identification of all hazards associated with the device, their causes, and the potential harm — including software-related hazards for devices incorporating software (which must also address IEC 62304 requirements).
Risk evaluation: Assessment of each risk against the defined acceptability criteria, before and after risk controls.
Risk controls: Documented implementation and verification of all risk control measures — both inherent safety by design, protective measures in the device, and information for safety.
Benefit-risk analysis: The central new requirement of EU MDR compared to MDD. Manufacturers must explicitly demonstrate that the clinical benefits of the device outweigh the residual risks — supported by clinical evidence. This is not a theoretical exercise: Notified Bodies expect a quantitative or semi-quantitative comparison of benefits and risks, referenced to clinical data.
Residual risk evaluation: Documentation of all residual risks after controls, with explicit confirmation that the overall residual risk is acceptable.
Section 5 — Product Verification and Validation
This section compiles all the technical testing evidence demonstrating that the device performs as intended and meets its specifications. It must include:
Pre-clinical testing: Biocompatibility testing per ISO 10993 series, electrical safety and EMC testing (IEC 60601 series for active devices), mechanical testing, software verification and validation (IEC 62304), usability engineering (IEC 62366), sterilization validation (where applicable), and shelf-life and packaging validation.
Biological safety is a core part of the verification and validation evidence: the Biological Evaluation Plan, chemical characterization reports and the Biological Evaluation Report all belong here. See our guide to biological evaluation under ISO 10993 for how this evidence is built and structured.
Software documentation: For devices incorporating software, the complete software lifecycle documentation per IEC 62304 — safety classification, software architecture, SOUP list, verification test records, and anomaly reports.
Clinical investigation: Where clinical investigation data exists, the full clinical investigation report and supporting data.
Equivalence: Where the manufacturer relies on data from equivalent devices rather than own clinical investigation, the technical, biological, and clinical equivalence argument under MDCG 2020-5, with access to the full technical documentation of the predicate device for Class III and implantable Class IIb devices.
Section 6 — Clinical Evaluation
The clinical evaluation is the most demanding and most frequently found-deficient section of EU MDR technical documentation. It must demonstrate, based on sufficient clinical evidence, that the device achieves its intended clinical benefits and that the identified risks are acceptable in the context of those benefits.
The clinical evaluation consists of:
Clinical Evaluation Plan (CEP): Defines the methodology, scope, and acceptance criteria for the clinical evaluation. It must be device-specific — generic templates are consistently flagged by Notified Bodies.
Clinical Evaluation Report (CER): The main document synthesizing all clinical evidence. It must cover the current knowledge/state of the art, demonstration of equivalence or own clinical data, appraisal and analysis of all relevant clinical data (literature, clinical investigation, post-market data), and clinical conclusions.
In the 2024 Team-NB survey, more than 30% of MDR non-conformities cited by Notified Bodies were linked to clinical evaluation or clinical evidence deficiencies — the single largest category of significant findings.
The most common CER deficiencies include: unclear safety and performance objectives that are vague or lack measurable acceptance criteria; inadequate state-of-the-art analysis; relying on competitor data for equivalence without documented access or clear comparability; and missing or insufficient clinical evidence without a documented justification or robust post-market plan.
Summary of Safety and Clinical Performance (SSCP): Required for Class III devices and implantable devices. A publicly available document summarizing the device’s safety and clinical performance data — validated by the Notified Body and published on EUDAMED.
Section 7 — Labelling and Instructions for Use
All labelling (device labels, packaging labels) and the Instructions for Use (IFU) must comply with MDR Annex I Chapter III (GSPR 23) and must be available in all official EU languages of the countries where the device is marketed. Common findings include missing mandatory label elements, IFU language coverage gaps, and inconsistencies between the IFU and the intended purpose as documented elsewhere in the technical file.
✦ PREMIUM BUNDLE · EU MDR COMPLETE
The complete EU MDR documentation set — technical, clinical and risk.
The Technical Documentation Kit (Annex II), the Clinical Documentation Kit (Articles 32, 61, 83-86) and the Risk Management Kit (ISO 14971 + IEC 62366-1) in one premium bundle. 22 templates, cross-referenced, with a single Master Index.
- ✓22 templates · pre-market + post-market documentation
- ✓Save €178 vs buying the three kits separately
Annex III — Post-Market Surveillance Technical Documentation
MDR Annex III defines the post-market surveillance technical documentation. Post-market surveillance under EU MDR is not an afterthought — it is a continuous, mandatory activity that feeds back into the technical documentation and the risk management file throughout the device’s commercial lifetime.
Post-Market Surveillance Plan (PMS Plan)
The PMS plan must be established before the device is placed on the market and must define the data sources to be monitored (complaint data, vigilance reports, EUDAMED, scientific literature, registries, post-market studies), the methods for collecting and analyzing that data, the frequency of data analysis, and the criteria that would trigger a corrective action.
Periodic Safety Update Report (PSUR) and Post-Market Surveillance Report (PMSR)
Notified bodies are examining post-market surveillance systems much more closely during EU MDR audits, and expectations around the PSUR and PMSR have tightened significantly.
The PSUR is required for Class IIa, IIb, and III devices. It must be updated at least annually for Class III and implantable Class IIb devices, and at least every two years for Class IIa and non-implantable Class IIb devices. The PSUR must include a summary of the device’s benefit-risk determination based on post-market data, conclusions from the PMS, and any corrective or preventive actions taken.
The PMSR is the equivalent document for Class I devices — less formal than the PSUR but still required to demonstrate active post-market monitoring
Post-Market Clinical Follow-Up (PMCF) Plan and Report
PMCF is the clinical component of post-market surveillance. The PMCF plan must define how the manufacturer will proactively collect and analyze clinical data after market release to confirm the device’s safety and performance, identify previously unknown risks, and address data gaps identified in the pre-market clinical evaluation.
PMCF is a frequent source of major findings in Notified Body audits — particularly when manufacturers submit generic PMCF plans that do not define specific data collection methods, timelines, and analysis criteria relevant to their specific device.
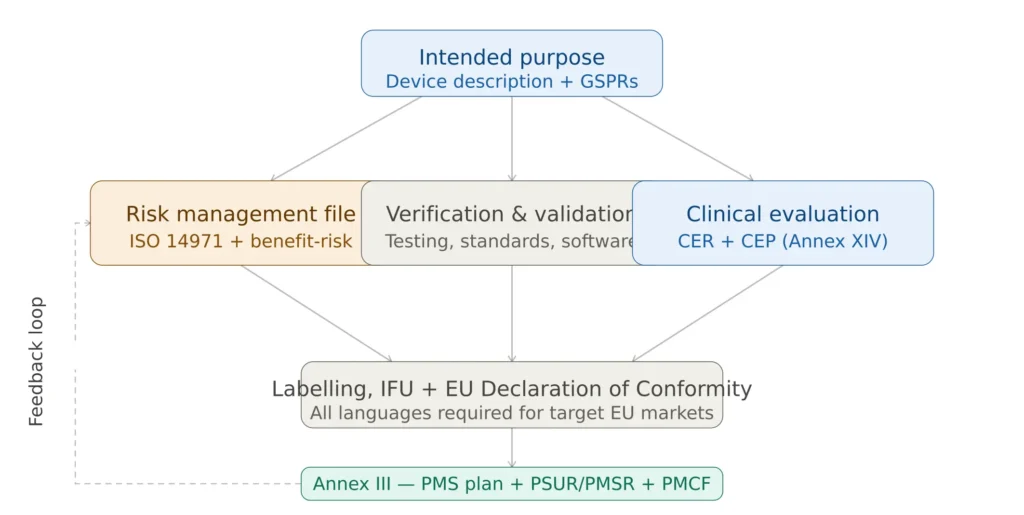
Figure 2 — EU MDR technical documentation traceability requirements
Key Differences Between MDR and MDD Technical Documentation
Understanding exactly how MDR raises the bar compared to the MDD is essential for manufacturers transitioning legacy devices and for those building documentation systems from scratch.
Clinical evidence depth: Under the MDD, clinical evaluation could often be satisfied with a relatively limited literature review and a generic equivalence claim. Under MDR, equivalence is substantially harder to claim — particularly for Class III and implantable devices, which require access to the full technical documentation of the predicate device, and where clinical equivalence must be demonstrated on the same patient population for the same indication. Stricter equivalence rules now require that technical, biological, and clinical similarity must be proven with access to complete technical documentation.
Benefit-risk analysis: The MDD required a risk analysis but did not explicitly mandate a benefit-risk balance. The MDR requires manufacturers to explicitly demonstrate, with reference to clinical evidence, that the clinical benefits outweigh the residual risks. This is a fundamentally different framing that changes how the entire technical file must be structured.
Post-market surveillance: The MDD’s post-market requirements were often satisfied with a reactive complaint handling system. MDR requires a proactive, structured PMS system with defined data sources, analysis methods, and update cycles — feeding back into the risk management file and clinical evaluation continuously.
Document coherence: Notified Bodies expect clear alignment between risk management, clinical evidence, labeling, and post-market activities. If those pieces don’t connect, findings usually follow. It helps to think of technical documentation as an interconnected system rather than a set of standalone reports.
Living documentation: Under the MDD, technical files could be relatively static. The MDR requires technical documentation to be kept up to date throughout the device’s lifecycle, updated when significant changes occur, and reviewed periodically even without changes.
Technical Documentation Requirements by Device Class
The depth and scope of documentation required scales with device risk classification, as summarized below.
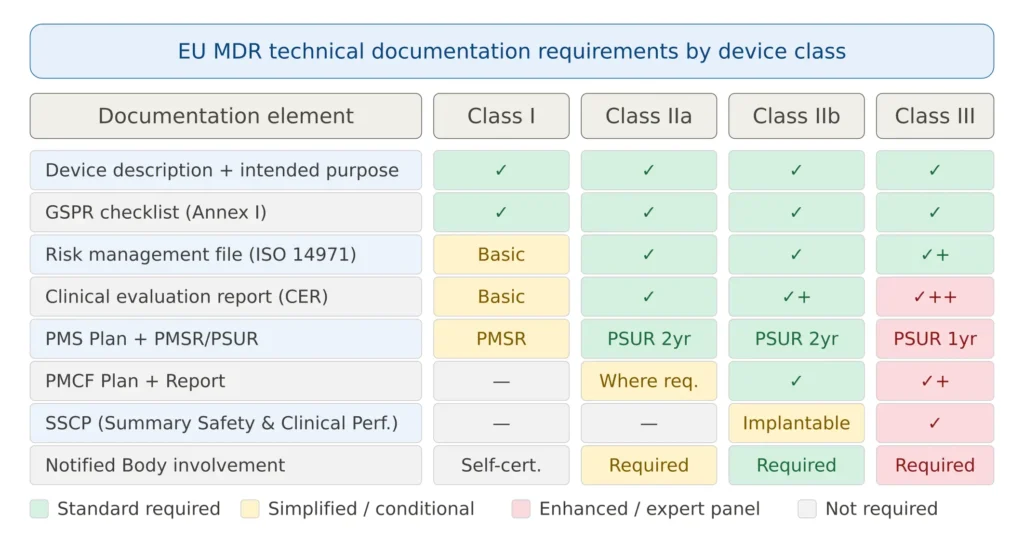
Figure 3 — EU MDR technical documentation requirements by device class
AUDIT-READY KIT
Build your ISO 13485 QMS with confidence.
Built on 15+ years of audit experience — every SOP and template references the regulations auditors expect. Get to certification faster, with industry best practices baked in.
- ✓30 SOPs covering the full QMS scope
- ✓56 templates ready to customize
- ✓Aligned with EU MDR + FDA QMSR
Common Notified Body Findings in Technical Documentation Reviews
Based on industry experience and published data from Notified Body surveys, the following are the most frequent findings during EU MDR technical documentation reviews. Addressing these proactively is the single most effective investment a manufacturer can make to accelerate Notified Body review timelines.
Vague or incomplete intended purpose: The intended purpose does not specify the medical condition, patient population, anatomical site, clinical setting, or operator qualifications with sufficient precision. A generic intended purpose statement — “the device is intended to diagnose and treat diseases” — does not meet MDR requirements.
Incoherent GSPR checklist: GSPRs are listed without adequate supporting evidence, or the standards referenced are outdated versions not currently harmonized. Many manufacturers also fail to document deviations from harmonized standards with appropriate justification.
Inadequate clinical evaluation: The most common issues include safety and performance objectives that are vague and lack measurable acceptance criteria; a state-of-the-art analysis that does not adequately address currently accepted methods; equivalence misuse by relying on competitor data without documented access; and gaps in clinical evidence without a documented plan or justification.
Documentation misalignment: A common issue is that the documents exist but they don’t tell a coherent story. Notified Bodies expect clear alignment between risk management, clinical evidence, labeling, and post-market activities. If those pieces don’t connect, findings usually follow.
Inadequate risk management integration: Risk management treated as a standalone document rather than integrated throughout the product lifecycle. The risk management file does not reference post-market data, and the benefit-risk analysis is not supported by clinical evidence.
Generic or template-based PMCF plan: PMCF plans that do not define specific data collection activities, timelines, acceptance criteria, or justification for why the selected approach will actually generate clinically meaningful data for this specific device.
Software documentation gaps: For devices incorporating software, incomplete IEC 62304 documentation — particularly missing SOUP lists, inadequate safety class justification, or absent traceability between software requirements and test cases.
Based on the EU Commission’s 2025 Economic Operator Survey, common submission issues include incomplete technical files, insufficient clinical evidence, inconsistent formatting, and unclear device intended use — all contributing to extended lead times during Notified Body review.
EU MDR Technical Documentation Table of Contents (IMDRF Structure)
Notified Bodies expect the technical file to follow a recognised table of contents. The IMDRF non-IVD Table of Contents (ToC) is the format most widely accepted across EU Notified Bodies, and it maps directly onto the Annex II and Annex III requirements described above. A typical EU MDR technical documentation table of contents is structured as follows:
- 1. Device description and specification — Annex II Section 1 (device description, variants, accessories, UDI, intended purpose)
- 2. Design and manufacturing information — Annex II Section 2 (design stages, manufacturing sites, sterilisation, supplier traceability)
- 3. General Safety and Performance Requirements checklist — Annex II Section 3 (GSPR matrix against Annex I, with evidence references)
- 4. Benefit-risk analysis and risk management — Annex II Section 4 (ISO 14971 risk management file, benefit-risk determination)
- 5. Product verification and validation — Annex II Section 5 (biocompatibility, electrical safety, software, usability, sterilisation, shelf-life)
- 6. Clinical evaluation — Annex II Section 6 (CEP, CER, SSCP where applicable)
- 7. Labelling and instructions for use — Annex II Section 7 (labels, IFU, language coverage)
- 8. Post-market surveillance documentation — Annex III (PMS plan, PSUR/PMSR, PMCF plan and report)
Building the technical file against this table of contents from the outset reduces completeness-check findings and gives the reviewer the familiar structure they expect. Manufacturers building from scratch can start from our ready-to-use EU MDR documentation kits, pre-structured to the IMDRF ToC and aligned to Annex II and III.
The Technical Documentation Review Process — What to Expect
Understanding how Notified Bodies actually review technical documentation helps manufacturers structure their documentation for maximum effectiveness.
According to Annex VII of the EU MDR, the Notified Body must assess technical documentation based on its predefined sampling plan, assess safety and performance requirements against Annex I, take into account requirements related to preclinical testing and clinical evaluations, and ensure that findings are appropriately and consistently classified based on a procedure to assess compliance with requirements of the EU MDR and relevant standards.
The review typically proceeds through three phases. First, a completeness check — verifying that all required sections are present and that the documentation follows a recognized structure (the IMDRF Table of Contents format is widely used). Second, a compliance assessment — evaluating whether the content actually demonstrates compliance with each MDR requirement. Third, a cross-referencing check — verifying consistency and coherence between sections.
According to the latest Notified Body survey results, it takes on average 13–18 months to issue a new EU certificate under the MDR or IVDR. However, the certification time depends not only on the availability of the Notified Body but also on how well the manufacturer has complied with the regulations in the first place.
Manufacturers who submit well-structured, coherent, and complete technical documentation consistently achieve faster review times — not because Notified Bodies process them differently, but because fewer questions and findings mean fewer response cycles.
Practical Guide: Building Your EU MDR Technical Documentation
Start with the intended purpose — and make it specific
Every other element of the technical documentation flows from the intended purpose. Invest significant time in defining it precisely before writing any other section. The intended purpose drives the device classification, the applicable GSPRs, the clinical evaluation scope, the risk analysis scope, and the PMS data requirements.
Structure your documentation around recognized frameworks
Use the IMDRF Table of Contents as your documentation structure. It is recognized by Notified Bodies across all EU markets and provides a common framework that reviewers are familiar with — reducing the risk of findings related to missing or misplaced sections.
Build traceability from the start
Create a traceability matrix that links every GSPR to its supporting evidence, every risk to its control measure and verification, and every clinical claim to its supporting clinical data. This matrix is the backbone of an audit-ready technical file and dramatically simplifies responses to Notified Body questions.
Treat the technical file as a living system
MDR compliance is not one document, one audit, or one Notified Body interaction — it is the outcome of a mature quality management system that consistently produces the right evidence. Establish a change control procedure that triggers technical documentation updates whenever the device design, manufacturing process, intended purpose, or labelling changes.
Engage your Notified Body early
Before formal submission, many Notified Bodies offer pre-submission meetings or preliminary document reviews. Use these opportunities to understand their specific expectations, identify potential gaps early, and build a productive working relationship.

Figure 4 — Common EU MDR technical documentation gaps and how to avoid them
AUDIT-READY KIT
Build your ISO 13485 QMS with confidence.
Built on 15+ years of audit experience — every SOP and template references the regulations auditors expect. Get to certification faster, with industry best practices baked in.
- ✓30 SOPs covering the full QMS scope
- ✓56 templates ready to customize
- ✓Aligned with EU MDR + FDA QMSR
Frequently Asked Questions
Do I need Annex II and III documentation for Class I self-certified devices? Yes — all devices require technical documentation per Annexes II and III, regardless of whether a Notified Body is involved. For Class I self-certified devices, you issue the EU Declaration of Conformity yourself and retain the technical documentation, but it must be complete and compliant. Competent authorities can request it at any time during market surveillance.
Can I use one technical documentation for multiple device variants? Yes — EU MDR allows a single technical documentation to cover a device family or range of variants, provided the variants share the same intended purpose, design principles, and manufacturing processes. Each variant must be individually listed with its specific UDI and any variant-specific characteristics documented.
How often must I update my technical documentation? Technical documentation must be updated whenever there is a change that could affect compliance — design changes, new clinical evidence, changes in applicable standards, post-market surveillance findings that require risk management updates. The PSUR (for Class IIa, IIb, III) triggers a structured periodic review at defined intervals (annually or bi-annually depending on device class). The documentation must also be kept up to date following annual Notified Body surveillance audits.
What is the difference between a technical file and technical documentation under EU MDR? These terms are essentially synonymous. “Technical file” was the term commonly used under the old MDD, while “technical documentation” is the precise term used in EU MDR 2017/745. The content requirements are defined in Annexes II and III of the Regulation.
Can I rely on data from an equivalent device to avoid clinical investigation? Equivalence is permitted under EU MDR, but the requirements are significantly stricter than under MDD. For Class III and implantable Class IIb devices, equivalence requires access to the full technical documentation of the predicate device — which is only practically feasible if the equivalent device belongs to the same manufacturer. For Class IIa and non-implantable Class IIb devices, equivalence is more achievable but must still be rigorously documented per MDCG 2020-5.
Conclusions
EU MDR technical documentation requirements under Annexes II and III represent a fundamental shift in how medical devices are regulated in Europe — from a documentation-focused compliance exercise to an evidence-based, continuously maintained demonstration of device safety and performance. The transition from MDD to MDR has raised the bar substantially, and the companies in the strongest position right now are using the transition period to improve documentation quality, close gaps in risk management and clinical evidence, strengthen post-market surveillance processes, and align their QMS fully with MDR and ISO 13485.
The key principles that define a successful EU MDR technical documentation system are coherence — every document must tell the same consistent story; traceability — every claim must be traceable to evidence; and continuity — the documentation must be actively maintained throughout the device’s commercial lifetime, not assembled once and left static.
For manufacturers still in the process of building or upgrading their technical documentation, the most important investment is a structured gap analysis against the Annex II and III requirements — ideally with reference to the Team-NB best practice guide and the relevant MDCG guidance documents — followed by a systematic remediation plan that prioritizes the highest-risk gaps first.
This article is part of the MD Regulatory series on EU MDR compliance. Related articles cover EU MDR transition deadlines 2026/2027, clinical evaluation requirements, post-market surveillance under EU MDR, and EUDAMED registration.