EU MDR GSPR Checklist: How to Demonstrate General Safety and Performance Requirements
Table of Contents
Every medical device placed on the EU market must demonstrate conformity with the General Safety and Performance Requirements set out in Annex I of Regulation (EU) 2017/745. The EU MDR GSPR requirements checklist is not an administrative formality — it is the structural backbone of the technical documentation, the document the Notified Body opens first, and the legal basis for the EU Declaration of Conformity. Every applicable requirement among the 23 articles of Annex I must be addressed with specific objective evidence; every non-applicable requirement must be justified with documented rationale. Gaps in the checklist propagate directly into Notified Body major non-conformities and CE certificate delays.
This guide is the practical companion for manufacturers building or revising their GSPR matrix. It walks through the structure of Annex I chapter by chapter, the demonstration logic for each major requirement, the role of harmonised standards and Common Specifications, the impact of the harmonisation gap that — as of 2026 — leaves the majority of requirements without a presumption-of-conformity standard, the cybersecurity expectations now firmly in place, and the structural format the GSPR checklist itself must take to survive Notified Body scrutiny. For the broader documentation context, see the EU MDR technical documentation guide and the EU MDR transition deadlines article for the regulatory timeline.
What the GSPR are and where they sit in Annex I
The General Safety and Performance Requirements replace the “Essential Requirements” of the previous Medical Device Directive 93/42/EEC. The change in name reflects a change in scope: the GSPRs are broader, more explicit on risk management, more demanding on clinical evidence, and substantially more prescriptive on labelling and information supplied with the device. They are organised across 23 numbered requirements split into three chapters.
Chapter I — General requirements (sections 1 to 9) establishes the foundational obligations. Section 1 requires that devices achieve their intended performance and be safe and effective. Section 2 introduces the obligation to reduce risks “as far as possible” without adversely affecting the benefit-risk ratio. Section 3 mandates a risk management system across the device lifecycle. Sections 4 to 9 cover risk control measures, performance during the device lifetime, packaging integrity, transport and storage, intended benefits versus residual risks, and the prohibition on devices whose risk-benefit balance is unacceptable. Chapter I applies to all devices without exception.
Chapter II — Requirements regarding design and manufacture (sections 10 to 22) applies selectively based on the device’s technological characteristics. Section 10 covers chemical, physical and biological properties. Section 11 covers infection and microbial contamination. Section 12 addresses devices incorporating substances considered medicinal products. Section 13 covers devices incorporating materials of biological origin. Section 14 addresses construction and interaction with the environment. Section 15 covers diagnostic and measuring functions. Section 16 covers protection against radiation. Section 17 — significantly expanded under MDR compared to the MDD — covers electronic programmable systems and software, including the cybersecurity and IT environment requirements. Section 18 covers active devices and devices connected to them. Section 19 covers particular requirements for active implantable devices. Section 20 covers protection against mechanical and thermal risks. Section 21 covers protection against risks posed to the patient or user by devices supplying energy or substances. Section 22 covers protection against risks posed by medical devices intended by the manufacturer for use by lay persons.
✦ PREMIUM BUNDLE · EU MDR COMPLETE
The complete EU MDR documentation set — technical, clinical and risk.
The Technical Documentation Kit (Annex II), the Clinical Documentation Kit (Articles 32, 61, 83-86) and the Risk Management Kit (ISO 14971 + IEC 62366-1) in one premium bundle. 22 templates, cross-referenced, with a single Master Index.
- ✓22 templates · pre-market + post-market documentation
- ✓Save €178 vs buying the three kits separately
Chapter III — Information supplied with the device (section 23) is a single but extensive requirement covering labelling and instructions for use. Section 23 specifies in detail what must appear on the label, on the packaging, and in the instructions for use, with specific provisions for sterile packaging, devices for lay use, and electronic instructions. Despite being one section, it is one of the most frequently audited and one of the most common sources of Notified Body findings.
The applicability rule is asymmetric. Chapter I and Chapter III apply to all devices. Chapter II applies in proportion to the device’s technological characteristics — a non-sterile mechanical device does not need to demonstrate conformity with section 11, but it must explicitly justify the non-applicability rather than skip the section silently.
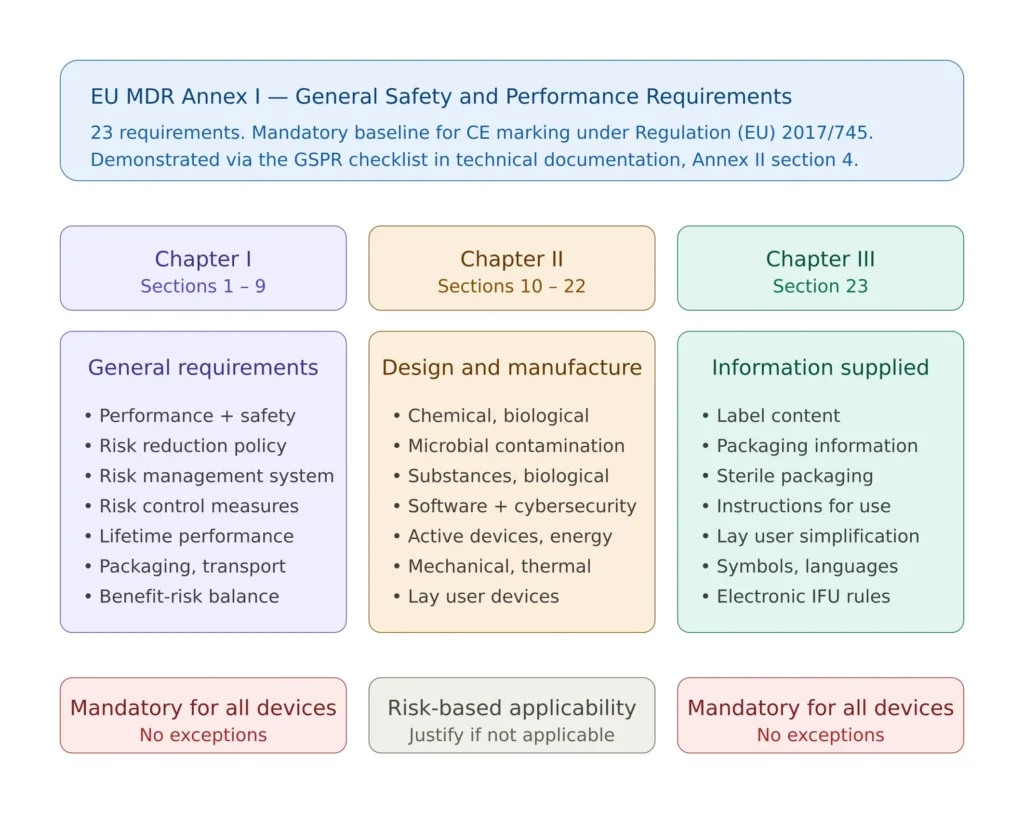
Figure 1 — Structure of EU MDR Annex I across the three chapters and the 23 requirements, showing what applies to all devices and what depends on technology.
Chapter I: the foundational requirements every device must address
Chapter I is where the GSPR checklist begins, and where Notified Body reviewers form their first impression of the manufacturer’s overall regulatory maturity. The nine sections of Chapter I are not aspirational principles — they are operational requirements that demand documented evidence.
Section 1 requires that devices achieve their intended performance and be safe and effective. The evidence here is the design verification record demonstrating that performance specifications are met under nominal conditions, supplemented by clinical evaluation per Article 61 demonstrating that the performance translates into clinical benefit. Performance claims in the technical documentation, on the label, and in marketing materials must be consistent with the verification and clinical evidence; an inconsistency here is a frequent finding.
Section 2 is the famous “as far as possible” obligation. Risks must be reduced as far as possible without adversely affecting the benefit-risk ratio, and the residual risks must be acceptable when weighed against the benefits. The phrase has been the source of considerable interpretation, but the operational reading is now stable: the manufacturer must demonstrate that risk reduction options were systematically considered, that those options yielding net benefit were implemented, and that those that would have degraded clinical performance or introduced larger risks were rejected with documented rationale. The MDCG guidance and Notified Body practice align on this point — “as far as possible” does not mean zero risk.
Section 3 requires a documented risk management system across the device lifecycle. Compliance is universally demonstrated through ISO 14971 risk management, which is harmonised under MDR. The risk management file feeds directly into multiple GSPR sections and is the most heavily cross-referenced piece of evidence in the GSPR checklist. Sections 4 to 8 elaborate the obligations on risk control measures, performance over the device lifetime, packaging integrity, transport conditions, and the explicit benefit-risk balance — each tied back to specific outputs of the ISO 14971 process.
Section 9 prohibits placing on the market devices whose risks cannot be reduced to an acceptable level. In the GSPR checklist, this requirement is normally addressed by reference to the benefit-risk analysis, which itself draws on the benefit-risk analysis methodology and outputs of the risk management process. A robust benefit-risk analysis is one of the highest-leverage documents in the entire technical file.
✦ EU MDR TECHNICAL DOCUMENTATION KIT
The complete Annex II technical file in one coordinated set.
8 Word templates covering the full EU MDR Annex II structure, plus Annex III (PMS) and Annex IV (Declaration of Conformity). Built around the latest MDCG guidance and Team-NB position papers. Suitable for all MDR classes (I, IIa, IIb, III) and custom-made devices.
- ✓8 templates · Annex II + III + IV
- ✓Team-NB position papers integrated as inline notes
Chapter II: design and manufacture requirements by technology
The thirteen sections of Chapter II apply selectively, but every section must be addressed in the GSPR checklist — either with evidence of conformity or with a justification of non-applicability. Skipping a section without justification is a process non-conformity regardless of whether the section was actually applicable.
Section 10 covers chemical, physical and biological properties of materials in contact with the patient or user. This is the entry point for the biocompatibility testing under ISO 10993 and for ISO 10993-1:2025, which provides the framework for the biological evaluation. For non-patient-contacting materials or devices with no biological evaluation requirement, the non-applicability must be justified with a clear rationale based on the device’s actual contact pattern.
Section 11 covers infection and microbial contamination. For sterile devices, the conformity demonstration relies on validated sterilisation per ISO 11135 (ethylene oxide), ISO 11137 (radiation), ISO 17665 (moist heat), ISO 14937 (general requirements) or other relevant standards. For non-sterile devices, the section is normally addressed through the bioburden controls embedded in the QMS and documented in the manufacturing controls section of the technical file.
Section 17 deserves particular attention because it has expanded significantly under MDR compared to the MDD and because it is the single most consequential section for software-containing devices and connected devices. Section 17.2 requires that software is developed and manufactured in accordance with the state of the art taking into account the principles of development lifecycle, risk management, including information security. Conformity is normally demonstrated through IEC 62304, and — for cybersecurity — through IEC 81001-5-1 and medical device cybersecurity testing. Section 17.4 explicitly requires manufacturers of software-controlled devices to set out minimum requirements concerning hardware, IT networks characteristics and IT security measures — including protection against unauthorised access — necessary to run the software as intended. This is the cybersecurity hook in MDR Annex I, and it has become a high-attention area in 2026 Notified Body audits.
Section 22 addresses devices intended for use by lay persons. Where a device is designed for use without professional supervision — home-use diagnostic devices, self-injection devices, wearables — the conformity evidence must specifically address lay user usability, typically through IEC 62366-1 usability engineering applied to a lay user population. Failure to apply IEC 62366 to a lay user device, or applying it to professional users only, is a recurring major finding.
Chapter III: the labelling and IFU requirements
Section 23 is one section but covers more text than several Chapter II sections combined. It specifies in detail what must appear on the label, on the packaging (with specific provisions for sterile packaging and packaging intended to maintain sterility), and in the instructions for use. The requirements are highly specific: the device’s name, the manufacturer’s name and address, the UDI, the device classification where applicable, the lot or serial number, the date of manufacture, the expiry date for relevant devices, indications and contraindications, warnings and precautions, residual risks communicated to the user, the intended user population, and many more elements depending on device type.
Two operational points dominate the practical compliance work on Section 23. First, the language requirement: information supplied with the device must be in the official language(s) of each Member State where the device is made available. For products marketed across the EU, this means up to 24 languages, with translation costs and version control implications. Second, the consistency requirement: every claim made on the label or in the IFU must be supported by evidence elsewhere in the technical file — claims without evidence are a finding, evidence without claims is a missed opportunity, and claims contradicting evidence is a critical finding.
For software-controlled devices and devices for which electronic instructions for use are permitted under Commission Regulation (EU) 2021/2226, the digital IFU obligations layer on top of Section 23. The conditions for using electronic IFU are specific — they apply mainly to professional use devices and to active implantables, with strict requirements on availability, archiving and the option for the user to request paper copy.
The harmonisation gap and what it means for compliance
Harmonised standards under Article 8 of the MDR provide a presumption of conformity with the GSPRs they cover. A manufacturer applying a harmonised standard correctly is presumed to comply with the corresponding GSPR — the burden of proof shifts. This presumption is the most efficient mechanism available for demonstrating compliance, and it is the path Notified Bodies expect manufacturers to take where harmonised standards exist.
The practical problem is that the harmonisation pipeline has lagged. As of January 2026, approximately 17% of the standards requested for harmonisation under MDR have been published in the Official Journal of the European Union — 48 of 277 requested standards. The remaining 83% sit in a regulatory grey zone where the standards exist and are widely used (ISO 14971, IEC 62304, ISO 10993 series, IEC 60601 series), but their use does not automatically trigger presumption of conformity. The full list of harmonised standards is maintained on the European Commission single market page and updated as new implementing decisions are adopted.
Three coping strategies are in use. First, applying the standard anyway and demonstrating its applicability through a documented gap analysis between the standard and the GSPR — this is the dominant approach and is accepted by Notified Bodies provided the demonstration is rigorous. Second, applying the harmonised version of an older directive standard (typically the MDD harmonised version) where the technical content is unchanged, with a justification of equivalence. Third, where Common Specifications exist, applying them — Common Specifications carry the same legal weight as harmonised standards and are mandatory unless equivalence is justified. Common Specifications have been adopted for specific high-risk device groups including reprocessing of single-use devices, some software-related cybersecurity expectations, and the device groups under Annex XVI of the MDR.
The implication for the GSPR checklist is structural: every requirement must be addressed with the explicit declaration of which standards are applied, whether those standards are harmonised, and what additional evidence is provided where the standards do not fully cover the GSPR. A checklist that lists “ISO 14971” without indicating its harmonised status under MDR (it is harmonised) and “IEC 62304” without flagging that it is not yet harmonised at the time of writing is technically incomplete.
AUDIT-READY KIT
Build your ISO 13485 QMS with confidence.
Built on 15+ years of audit experience — every SOP and template references the regulations auditors expect. Get to certification faster, with industry best practices baked in.
- ✓30 SOPs covering the full QMS scope
- ✓56 templates ready to customize
- ✓Aligned with EU MDR + FDA QMSR
Building the GSPR checklist that survives Notified Body review
The GSPR checklist is located in section 4 of the technical documentation under MDR Annex II. Notified Bodies expect a specific structural format with five required elements per requirement. A checklist missing any of these five elements will trigger comment or non-conformity regardless of how strong the underlying evidence is.
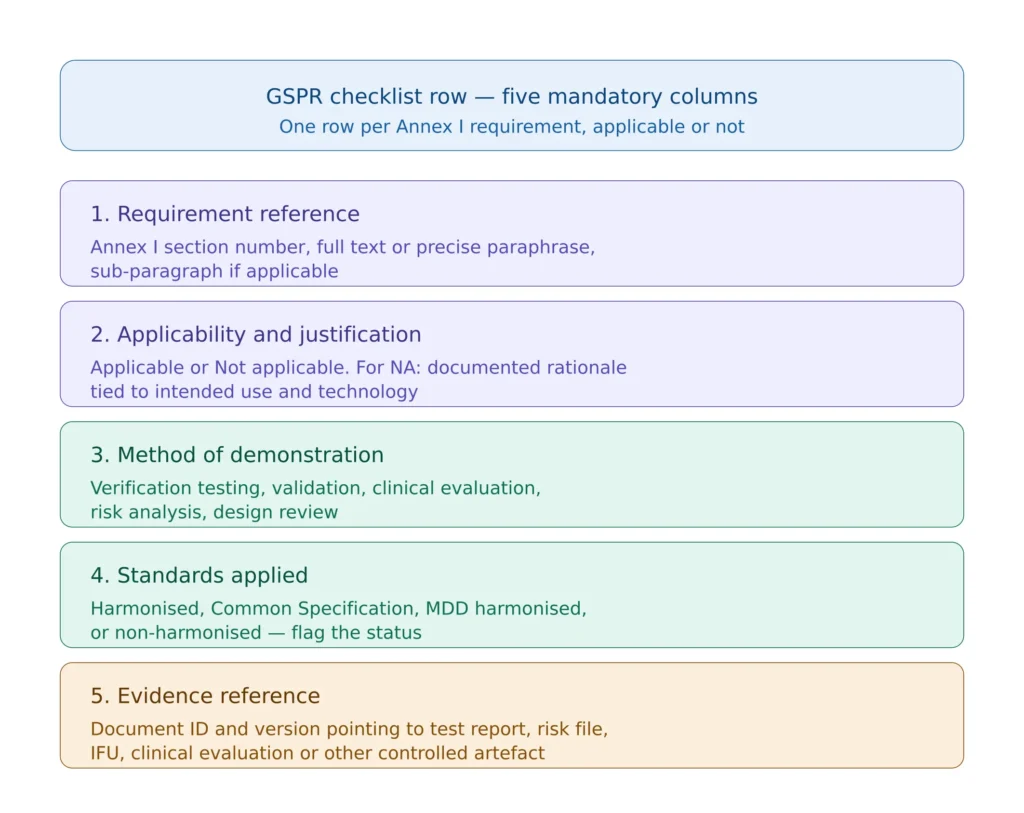
Figure 2 — The five mandatory columns of a GSPR checklist that meets MDR Annex II expectations.
The first column is the requirement reference itself — the section number from Annex I, the full requirement text or a precise paraphrase, and any relevant sub-paragraph. Some manufacturers use the full Annex I text verbatim; others use a precise paraphrase to keep the matrix compact. Both are acceptable provided the paraphrase does not lose content. What is not acceptable is a numerical reference without text — the auditor must be able to read the requirement and the response side by side.
The second column records applicability. “Applicable” is a single word; “Not applicable” requires justification in the same row, with rationale tied to the device’s intended use and technology. Justifications such as “device is not active” for section 18, or “device does not contain materials of biological origin” for section 13, are sufficient when factually correct. Justifications that conflate sections, that miss applicable requirements through over-narrow reading, or that simply state “N/A” without text are findings.
The third column declares the method of demonstration. The methods are limited in number and include design verification testing, validation, clinical evaluation, risk analysis output, design review, manufacturing process control, and labelling content review. The method must be appropriate to the requirement — a clinical evaluation cannot demonstrate sterilisation efficacy, and bench testing cannot demonstrate clinical benefit.
The fourth column lists the standards applied with their harmonised status. The format used in mature checklists is “ISO 14971:2019 (harmonised under MDR — Implementing Decision (EU) 2024/2336)” or “IEC 62304:2006/A1:2015 (not harmonised under MDR; applied as state of the art)” or “Common Specification per Implementing Regulation (EU) 2022/2347”. This level of explicitness is what differentiates a defensible checklist from a tickbox checklist.
The fifth column references the evidence with document ID and version. The reference must point to a controlled document in the QMS that the auditor can request and review. Generic references such as “design documentation” or “test reports” are findings — the auditor expects a specific document number, version, and ideally the section or page within that document where the evidence sits.
A sixth column is added in some implementations to record the assessment of the assigned reviewer, the date of last review, and the post-market surveillance feedback that may have updated the conformity status. This is good practice though not strictly mandatory.
Cybersecurity expectations under section 17
In 2026 cybersecurity is no longer a niche technical topic in MDR conformity demonstration — it is a core regulatory expectation that Notified Bodies probe explicitly during technical documentation review. Section 17.2 requires software development to address information security; Section 17.4 requires manufacturers to set out minimum requirements for hardware, IT networks and IT security measures. The MDCG 2019-16 guidance on cybersecurity for medical devices, originally published in 2019 and updated since, defines the operational expectations.
The conformity demonstration for cybersecurity-relevant aspects of section 17 typically draws on multiple inputs: the security risk management performed under IEC 81001-5-1 (which integrates with the device-level ISO 14971 risk management); the secure development lifecycle evidence under IEC 62304 Section 17 alignment; the cybersecurity testing evidence (vulnerability scanning, penetration testing, fuzz testing where applicable) per the medical device cybersecurity testing framework; and the labelling and IFU content describing the IT environment requirements, security configuration responsibilities of the operator, and the manufacturer’s vulnerability disclosure and patch policy.
A cybersecurity-relevant device whose GSPR checklist addresses section 17 with a single line referencing IEC 62304 — without IEC 81001-5-1, without testing evidence, and without IFU content — is the most common configuration found in technical documentation and the most common source of major non-conformity at first MDR submission. The Software as a Medical Device regulatory framework adds further specific expectations for SaMD devices that must be reflected in the GSPR checklist.
Cross-references that hold the technical file together
The GSPR checklist is not an island. It is the cross-reference document that connects the rest of the technical file. Each evidence reference in column five points to a controlled artefact, and each artefact must in turn be internally consistent.
The clinical evaluation report referenced under sections 1, 2, 8 and others must be consistent with the intended purpose declared in section 1, with the residual risks accepted in section 9, and with the claims made in the IFU under section 23. The risk management file referenced under section 3 must trace its hazards through to the labelling warnings under section 23. The biocompatibility evaluation under section 10 must align with the materials listed in the design documentation, the manufacturing controls, and the IFU statements about contraindications.
These cross-consistency checks are exactly what Notified Body reviewers perform first. A checklist that internally appears robust but contradicts the clinical evaluation, the risk file, or the IFU is worse than a sparse but consistent checklist — the inconsistency is interpreted as evidence that the manufacturer does not have control of the technical file.
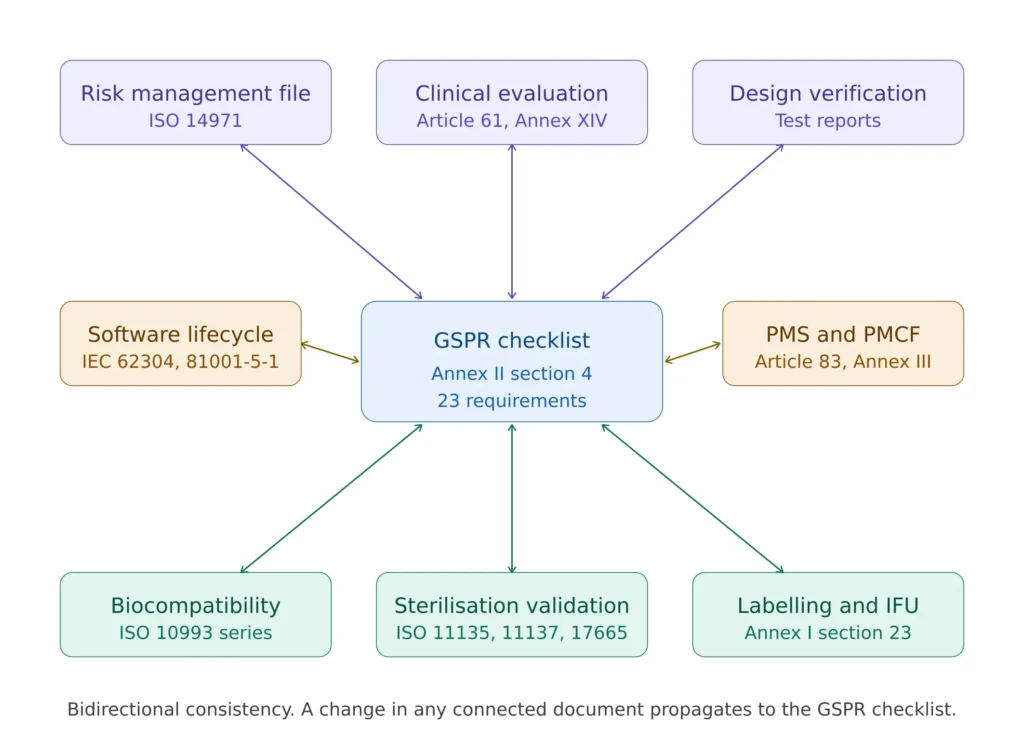
Figure 3 — How the GSPR checklist connects to the rest of the MDR technical documentation under Annex II.
Common audit findings on GSPR demonstration
Across MDR technical documentation reviews and Notified Body audits, a recognisable set of GSPR-related findings recurs. Each is preventable at the checklist preparation stage if the structural discipline is in place from the start.
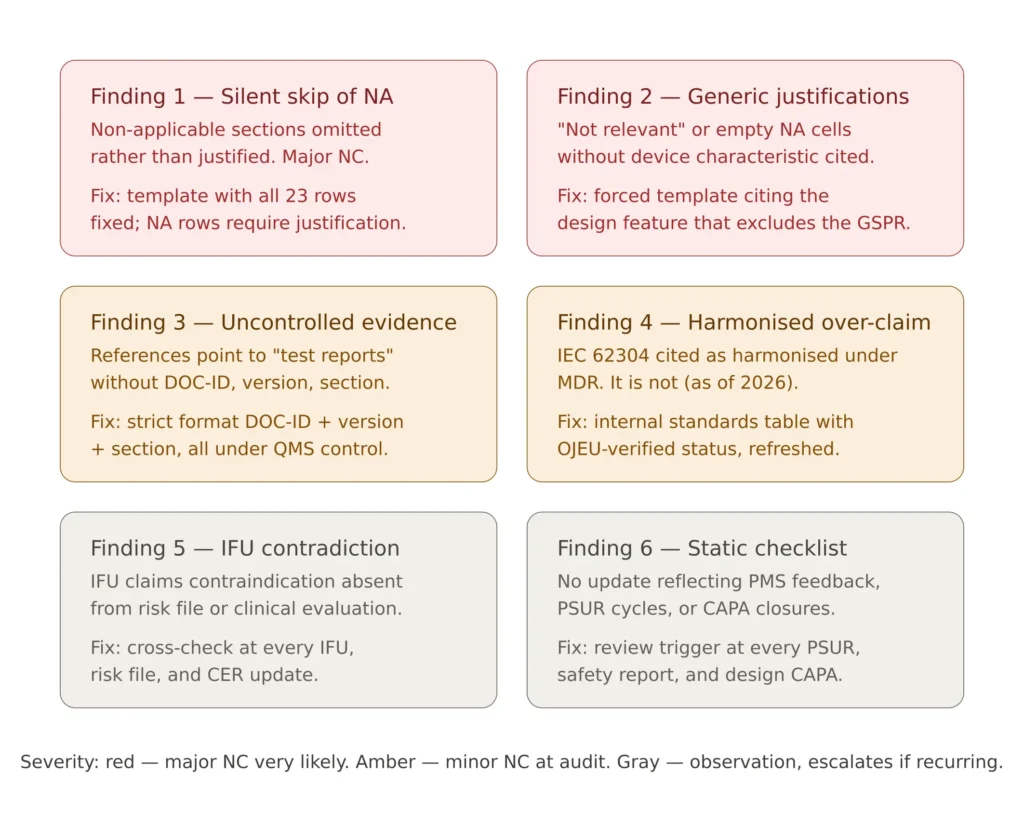
Figure 4 — The six most common GSPR checklist audit findings, with the practical fix for each.
The first recurring finding is the silent skip of non-applicable requirements. A checklist that contains rows for Chapter II sections 10 to 22 only where they apply, with the non-applicable sections simply omitted, is incomplete. Annex II requires the demonstration to include all GSPRs that apply and all those that do not, with justification for the latter. The fix is structural: the checklist template includes all 23 requirements as fixed rows; non-applicable ones receive a justification rather than a deletion.
The second is generic non-applicability justifications. “Not applicable” without text, or with text such as “not relevant to this device”, is a finding regardless of whether the requirement actually applies. The fix is a justification template that forces the writer to state the device characteristic that excludes the requirement: “Not applicable. The device does not contain materials of biological origin (no animal-derived, human-derived or microbial-derived components in the design BOM, ref DOC-BOM-001)”.
The third is evidence references to non-existent or non-controlled documents. A checklist column-five entry pointing to “Risk Management Plan” without document number, version or location is uncontrollable. The fix is a strict format: every evidence reference is “DOC-ID, version, section” or equivalent, pointing to an artefact under the QMS.
The fourth is over-claiming on harmonised standards. A checklist that states “ISO 14971:2019 (harmonised)” for every section that mentions risk, or “IEC 62304:2006 (harmonised)” without checking the standard’s harmonised status under MDR, propagates errors that will be caught. The fix is a maintained internal table of standards used by the organisation, with their MDR harmonised status verified against the OJEU and updated on a defined cycle.
The fifth is contradiction between the GSPR checklist and the IFU or label content. The IFU claims a contraindication that is not reflected in the risk management file; the label declares an environmental condition not supported by the design verification; the IFU describes a use scenario the clinical evaluation does not cover. The fix is a controlled cross-check at every checklist update: when the IFU changes, the checklist is reviewed; when the risk file is updated, the checklist is reviewed.
The sixth is the static checklist — a checklist updated only at major design changes, with no reflection of post-market surveillance feedback under EU MDR post-market surveillance. PMS feedback that affects the benefit-risk balance, that introduces new risk control measures, or that updates the residual risk assessment must propagate to the checklist. The fix is a documented review trigger: the checklist is reviewed at every PSUR cycle, every safety report, and every CAPA closure that affects design or labelling.
State of the art and the 2026 cybersecurity bar
The phrase “state of the art” appears repeatedly in Annex I and is one of the most frequently misunderstood concepts in MDR conformity demonstration. State of the art is not a synonym for “the most advanced technology”; it is the developed stage of technical capability at a given time, including the available scientific, technical and clinical knowledge, and including the consensus of expert opinion as embodied in published standards, guidance and practice.
Operationally, state of the art is demonstrated by reference to current versions of standards, current guidance documents (MDCG documents, FDA guidances where relevant), recent clinical literature, and industry best practice as reflected in trade association outputs. The state of the art evolves; a GSPR checklist prepared three years ago referencing standards that have since been superseded — even if the older versions were used during design — is technically defensible only if the gap has been assessed and the residual risk found acceptable.
Cybersecurity is the area where state of the art has moved most rapidly. The 2026 expectations under section 17.4 reflect a developed practice that did not exist in 2017 when the MDR was adopted: secure software development lifecycle aligned to IEC 81001-5-1, vulnerability management with documented disclosure policy, threat modelling integrated with the device-level risk management, penetration testing for connected devices, and explicit communication to operators of the IT environment requirements and the boundaries of manufacturer responsibility. A GSPR checklist for a connected device that does not address any of these elements under section 17 is not at the state of the art and will be challenged.
Frequently asked questions
What are the EU MDR GSPR requirements and where are they defined?
The General Safety and Performance Requirements are defined in Annex I of Regulation (EU) 2017/745 — the Medical Device Regulation. They consist of 23 requirements organised in three chapters: Chapter I sections 1 to 9 cover general requirements, Chapter II sections 10 to 22 cover design and manufacture requirements that apply based on technology, and Chapter III section 23 covers information supplied with the device including labelling and instructions for use. Chapter I and Chapter III apply to all devices; Chapter II applies selectively with documented justification for non-applicable sections.
Is a GSPR checklist mandatory under EU MDR?
Yes, in practice. Annex II section 4 of the MDR requires the technical documentation to contain a structured demonstration of conformity with the applicable GSPRs, and Notified Bodies expect this demonstration in checklist or matrix format covering all 23 requirements. The checklist must include the requirement reference, applicability and justification, method of demonstration, standards applied, and evidence references for each row. A technical file that addresses GSPRs in narrative form without a structured matrix will be challenged.
What is the difference between a harmonised standard and a Common Specification?
Harmonised standards are European standards developed by CEN, CENELEC or ETSI on the basis of a Commission mandate, with their references published in the Official Journal of the European Union. Their use is voluntary but provides a presumption of conformity with the GSPRs they cover. Common Specifications are technical specifications adopted by Commission Implementing Regulation when no harmonised standard exists or when existing standards are insufficient. Common Specifications carry the same legal weight as harmonised standards and are mandatory unless the manufacturer can justify equivalent solutions.
Why are so few standards harmonised under MDR?
As of January 2026, approximately 17% of the standards requested for harmonisation under MDR have been published in the Official Journal — 48 of 277. The harmonisation process is slow due to procedural complexity in the EU standardisation request mechanism and the technical content review by Commission consultants. The practical effect is that most widely-used standards (IEC 62304, ISO 14971, ISO 10993 series, IEC 60601 family) are applied as state of the art rather than under formal presumption of conformity. Notified Bodies accept this approach provided the application of the standard to the GSPR is demonstrated in the checklist.
How do I justify non-applicability of a GSPR?
The justification must reference a specific characteristic of the device that excludes the requirement, tied to the intended use and design. Examples of acceptable justifications: “Not applicable — device is not active and contains no source of energy, ref DOC-DESIGN-001 section 3.2” for section 18; “Not applicable — device is supplied non-sterile and has no microbial control claims, ref DOC-IFU-001” for section 11.6 on sterile packaging. Generic justifications such as “not relevant” or unwritten “N/A” entries are findings.
What documents must the GSPR checklist reference?
Each applicable GSPR row must point to specific controlled documents. Typical references include the risk management file (ISO 14971), clinical evaluation report (Article 61, Annex XIV), design verification reports, biocompatibility evaluation (ISO 10993 series), sterilisation validation reports, software lifecycle documentation (IEC 62304, IEC 81001-5-1 for cybersecurity), labelling and IFU documents, and post-market surveillance plan and PSUR. References must include document ID and version, not generic descriptions.
How often must the GSPR checklist be updated?
The checklist is a living document. It must be updated at every design change that affects an applicable requirement, every change in the IFU or labelling, every change in the risk management file that affects residual risk acceptability, and every PSUR cycle where post-market data introduces new information. As a minimum, an annual review is good practice. Notified Bodies expect the checklist version date to be recent relative to the technical documentation submission; a checklist last updated three years before a Notified Body review is presumptively stale.
Does the GSPR checklist apply to legacy MDD devices transitioning to MDR?
Yes. A device transitioning from MDD certification to MDR must produce a full GSPR checklist as part of the MDR technical documentation, even when the design is unchanged from its MDD configuration. The transition timeline is set by the MDR transition deadlines, and the GSPR checklist is one of the most substantial pieces of new documentation that legacy devices must produce — the MDD Essential Requirements checklist does not map one-to-one to the MDR GSPR matrix and must be rebuilt rather than translated.
Conclusions
The GSPR checklist is the single most leveraged document in the MDR technical documentation. It is the cross-reference that connects clinical evaluation, risk management, design verification, biocompatibility, sterilisation validation, software lifecycle and labelling into a coherent demonstration of conformity. A well-structured checklist makes a Notified Body review efficient and predictable; a poorly-structured one makes the entire technical file appear unreliable regardless of how strong the underlying evidence is.
The five-column structural discipline — requirement reference, applicability with justification, method of demonstration, standards applied with harmonised status, evidence reference with document ID and version — is the format Notified Bodies expect and the format that survives audit. The 2026 cybersecurity bar under section 17 is now firmly in place and must be addressed with concrete IEC 81001-5-1 alignment and testing evidence rather than a passing reference to IEC 62304. The 83% harmonisation gap means most evidence is presented as state-of-the-art application rather than under presumption of conformity, and the checklist must be explicit about which standards carry the presumption and which do not.
The EU MDR Technical Documentation Package on MD Regulatory includes the complete GSPR checklist template covering all 23 Annex I requirements with structured columns, justification templates for non-applicability, the standards reference table with MDR harmonised status, the cross-reference map to risk management and clinical evaluation, the cybersecurity-specific section 17 evidence template, and the technical documentation index aligned to MDR Annex II — all immediately deployable in an existing ISO 13485 quality management system and ready for Notified Body submission.
For the broader technical documentation context, see the EU MDR technical documentation guide. For the clinical evidence requirements that feed multiple GSPR sections, the EU MDR clinical evaluation framework under Article 61 and Annex XIV is the complementary reading. For the foundational risk management framework that connects to Chapter I sections 3, 4 and 8, see ISO 14971 risk management.