ISO 13485 Design Controls: Requirements and Design History File
Table of Contents
The ISO 13485 design control requirements are where most small and medium medical device manufacturers fail their first certification audit. Clause 7.3 of ISO 13485:2016 governs how a device is planned, specified, verified, validated, transferred to production and changed over its life. It is also the single most heavily sampled area in any Notified Body or regulatory audit, because it is the part of the quality system that most directly determines whether a device is safe and performs as intended. The Design History File (DHF) — the complete record proving the design was actually controlled — is the artefact auditors ask for first, and the artefact that is most often incomplete.
This guide explains the ISO 13485 design control requirements clause by clause: planning, design inputs and outputs, design review, verification, validation, transfer and change control, and how all of these accumulate into a defensible DHF. It is written for quality and regulatory professionals who already operate a quality management system and need to make Clause 7.3 audit-proof rather than merely documented. Throughout, design controls are treated as part of the wider system described in our ISO 13485 complete guide, and the risk-management connections are explored in depth in our ISO 14971 risk management article.
Why ISO 13485 Design Controls Exist
Design controls are not bureaucratic overhead. They exist because the overwhelming majority of field failures and recalls do not originate in manufacturing — they originate in design. A device that is correctly built to a flawed specification is still a dangerous device. Clause 7.3 forces the organisation to convert subjective intentions (“the device should be easy to use and safe”) into objective, testable requirements, and then to prove, with retained records, that the finished design actually meets them.
AUDIT-READY KIT
Build your ISO 13485 QMS with confidence.
Built on 15+ years of audit experience — every SOP and template references the regulations auditors expect. Get to certification faster, with industry best practices baked in.
- ✓30 SOPs covering the full QMS scope
- ✓56 templates ready to customize
- ✓Aligned with EU MDR + FDA QMSR
ISO 13485:2016 remains the current edition. The ISO technical committee TC 210 conducted a systematic review and reconfirmed the 2016 edition rather than revising it, prioritising stability. This stability matters because the FDA’s Quality Management System Regulation (QMSR) became effective on 2 February 2026 and now incorporates ISO 13485:2016 by reference, replacing the design controls language formerly at 21 CFR 820.30, as confirmed in the FDA Quality Management System Regulation. A manufacturer that builds its design control system around Clause 7.3 today is therefore building to a requirement that is stable and that simultaneously satisfies both EU and US expectations. The detail of how design evidence flows into the EU technical file is covered in our EU MDR technical documentation guide.
The remainder of this article walks through the lifecycle in the order an auditor reasons about it. Figure 1 shows the complete Clause 7.3 lifecycle and, critically, how every activity feeds the Design History File.
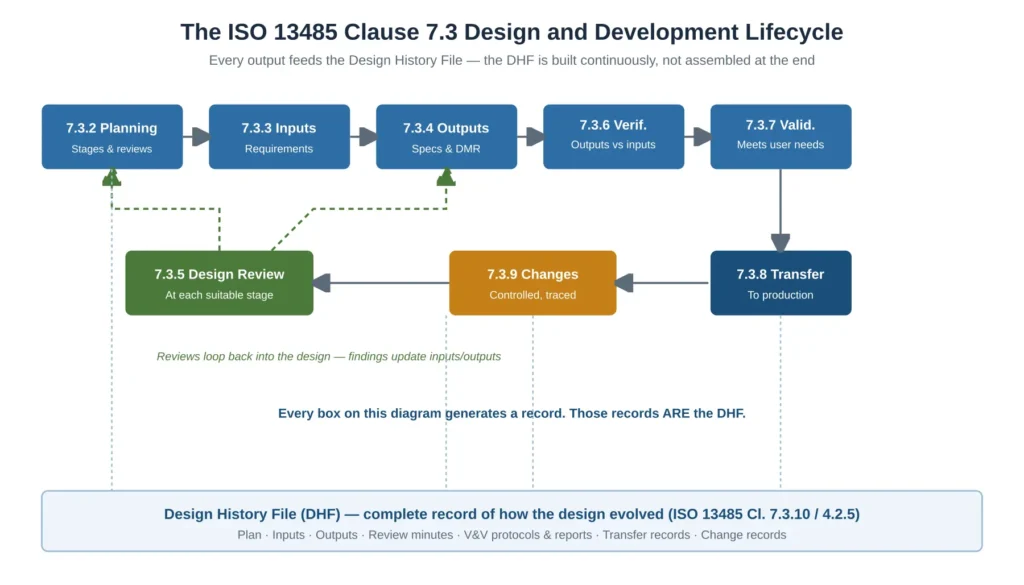
Figure 1 — The ISO 13485 Clause 7.3 design and development lifecycle and how each stage feeds the Design History File
Design and Development Planning (Clause 7.3.2)
Planning is the requirement most often treated as a one-off document and most often cited as a finding. Clause 7.3.2 requires the organisation to plan and control the design, and to keep that plan updated as the design progresses. The plan must define the design and development stages, the review, verification, validation and transfer activities appropriate at each stage, the responsibilities and authorities, the methods to ensure traceability, and the resources needed including necessary competence.
The error auditors look for is not the absence of a plan but its fossilisation: a plan written at project kick-off, approved, and never touched again while the project changed substantially around it. When the plan no longer reflects reality, every downstream record becomes questionable. The practical fix is to treat the design plan as a living controlled document, revised and re-approved at each design review milestone, with a revision history that demonstrates it tracked the actual project. Planning also explicitly includes planning the interfaces between different groups involved in design — a requirement that becomes a real finding when software, hardware, and regulatory teams work from divergent assumptions.
Design Inputs and Design Outputs (Clauses 7.3.3 and 7.3.4)
Inputs and outputs are the load-bearing structure of the entire clause. Everything that follows — verification, validation, transfer, change control — is meaningless unless inputs and outputs are correctly defined and traceable to each other. Figure 2 summarises the requirement, the mandatory record, and the typical audit failure for each sub-clause discussed in this article.
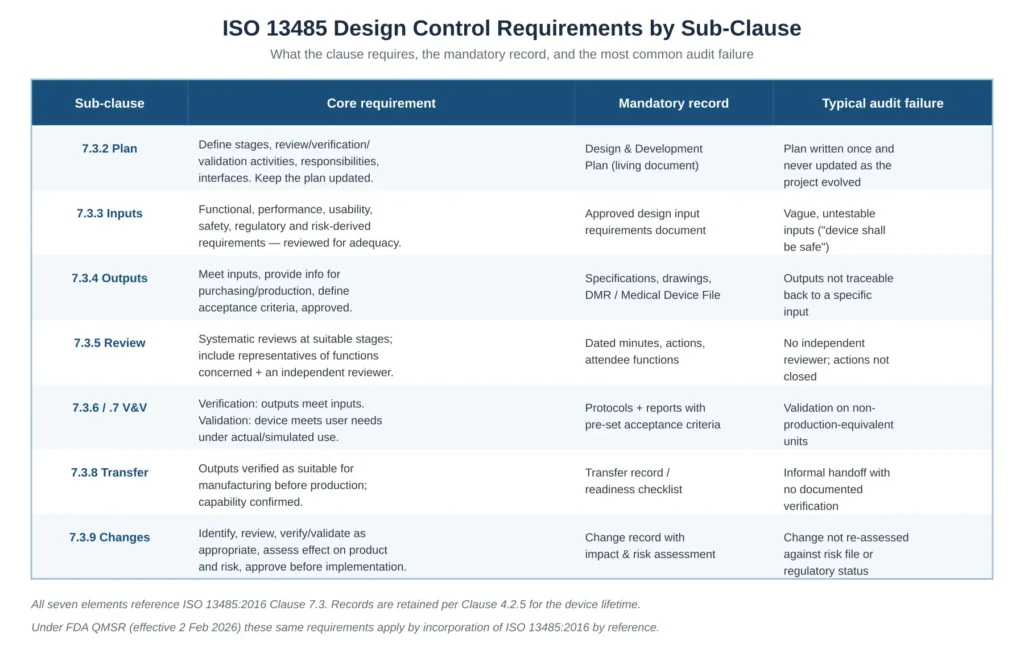
Figure 2 — ISO 13485 design control requirements by sub-clause, with the mandatory record and the typical audit failure for each
Design Inputs (Clause 7.3.3)
Design inputs are the requirements the device must meet. Clause 7.3.3 requires them to include functional, performance, usability and safety requirements according to the intended use; applicable regulatory requirements and standards; the outputs of risk management; and, where appropriate, information derived from previous similar designs. Inputs must be reviewed for adequacy and approved, and they must be complete, unambiguous, and capable of being verified or validated.
✦ PREMIUM BUNDLE
The ultimate global QMS documentation bundle.
Combine ISO 13485 + all 5 MDSAP markets in one premium package. Deduplicated structure means you customize each document once — not twice. Save €199 vs buying the kits separately.
- ✓41 SOPs covering both ISO 13485 and MDSAP
- ✓70+ templates with deduplicated structure
- ✓Save €199 vs buying separately
The single most common input failure is the untestable requirement. “The device shall be safe” cannot be verified; “the enclosure shall withstand a 1.0 m drop onto concrete on each face without loss of function” can. Risk management is an explicit input source: every risk control measure identified under ISO 14971 must appear as a design input so that it can be implemented in an output and proven in verification. This is the mechanism by which the risk file and the design file stay coupled, and a broken coupling here is one of the most serious findings an auditor can raise. The relationship between risk controls and benefit-risk justification is developed further in our benefit-risk analysis article.
Design Outputs (Clause 7.3.4)
Design outputs are the results of the design effort expressed in a form that can be verified against the inputs. Clause 7.3.4 requires outputs to meet the input requirements, to provide appropriate information for purchasing, production and service provision, to contain or reference acceptance criteria, and to specify the characteristics of the product essential for its safe and proper use. Outputs are approved before release.
In practice the outputs are the specifications, drawings, software, and the Medical Device File content used for production — what FDA historically called the Device Master Record. The decisive test an auditor applies is bidirectional traceability: can every output be traced back to a specific input, and can every input be traced forward to an output that satisfies it? An output with no parent input suggests uncontrolled scope creep; an input with no child output suggests a requirement that was silently dropped. For software devices, the structure of outputs is governed in parallel by IEC 62304, as covered in our IEC 62304 complete guide.
Design Review (Clause 7.3.5)
Clause 7.3.5 requires systematic design reviews at suitable stages in accordance with planned arrangements. Reviews must evaluate the ability of the design to meet requirements, identify problems, and propose necessary actions. A specific structural requirement is frequently missed: the participants must include representatives of functions concerned with the design stage being reviewed, plus other specialist personnel — and the standard’s intent, reinforced by auditor practice, is that at least one reviewer is independent of the design activity being reviewed so the review is not the designers marking their own work.
The records must identify the design under review, the participants and their functions, the date, and the results including any actions and their owners. The most common review finding is not the absence of reviews but the absence of independence and the absence of action closure: meetings were held, but actions raised were never tracked to completion, leaving the review evidentially worthless. Reviews are also the formal mechanism through which the design loops back on itself, as shown by the feedback path in Figure 1 — findings update inputs and outputs rather than being noted and forgotten.
Design Verification and Design Validation (Clauses 7.3.6 and 7.3.7)
Verification and validation answer two different questions and conflating them is one of the most damaging errors in a design file. Verification asks: did we build the device right — do the outputs meet the inputs? Validation asks: did we build the right device — does it meet the user’s needs and intended use in real or simulated conditions? A device can pass every verification test and still fail validation if the inputs themselves were wrong.
Design Verification (Clause 7.3.6)
Clause 7.3.6 requires verification to confirm that design outputs meet design inputs, in accordance with planned arrangements, with documented protocols that include acceptance criteria defined before testing begins and, where appropriate, statistical techniques with a rationale for sample size. The records must show what was tested, against which input, by which method, and with what result. Acceptance criteria defined after seeing the data is a finding, because it allows the result to define the requirement rather than the requirement governing the result.
Biological safety is a core part of the verification and validation evidence: the Biological Evaluation Plan, chemical characterization reports and the Biological Evaluation Report all belong here. See our guide to biological evaluation under ISO 10993 for how this evidence is built and structured.
Design Validation (Clause 7.3.7)
Clause 7.3.7 requires validation to ensure the resulting product is capable of meeting the requirements for the specified application or intended use. Validation must be performed on representative product — production units, batches, or their equivalents — and the rationale for what was used must be recorded. Validation includes usability and, where relevant, clinical evaluation. The classic finding is validation performed on engineering prototypes that differ materially from production product, which proves nothing about what the patient will actually receive. Usability validation links to IEC 62366, and for software the verification and validation split is examined in detail in our IEC 62304 software verificationguide. Note carefully that IEC 62304 covers software verification only; software validation derives from IEC 82304-1, the ISO 13485 design controls described here, and IEC 62366 usability — these are distinct and must not be conflated in any software design file.
Design Transfer and Design Changes (Clauses 7.3.8 and 7.3.9)
Clause 7.3.8 requires procedures for transfer of design and development outputs to manufacturing. The outputs must be verified as suitable for manufacturing before they become production specifications, and the results of that verification recorded. Under the FDA QMSR, design transfer is now governed by this ISO clause rather than the former 21 CFR 820.30(h), and informal handoffs that assume manufacturability are explicitly insufficient. The finding here is the undocumented handoff: production simply “starts” without a recorded confirmation that the outputs are actually producible at scale and that production specifications faithfully reflect the approved design.
Clause 7.3.9 requires that design changes are identified, reviewed, verified, validated as appropriate, and approved before implementation. The review of changes must include evaluation of the effect of the change on constituent parts and product already delivered, and — critically — on the inputs and outputs of risk management. The most serious change-control finding is a post-launch change that was implemented without re-assessing the risk file, the verification evidence, the labelling and claims, and the regulatory status, including whether the change is a significant change requiring notification. Records of changes and the actions arising must be maintained. The discipline required here is essentially the same as for corrective action, and integrates with the system described in our ISO 13485 CAPA procedure guide.
The Design History File: Structure and Traceability
ISO 13485:2016 requires, in Clause 7.3.10, that the organisation maintain a design and development file for each medical device type, containing or referencing the records generated by the design process. This file is what the industry continues to call the Design History File. Records are retained for at least the lifetime of the device as defined by the organisation, but not less than the retention period of any resulting record or as specified by applicable regulatory requirements, consistent with Clause 4.2.5.
A compliant DHF is not a folder of loose files. It is an index that lets an auditor select any requirement and walk it end to end: from a user need, to the design input that captured it, to the output that implemented it, to the verification that confirmed the output meets the input, to the validation that confirmed the user need is met, to the risk control it satisfies. Figure 3 shows both the DHF index structure and that traceability chain. The traceability matrix is the spine of the file; a single broken link in it is the most common Clause 7.3 nonconformity raised in SME audits, and the reason DHFs are so frequently judged incomplete.
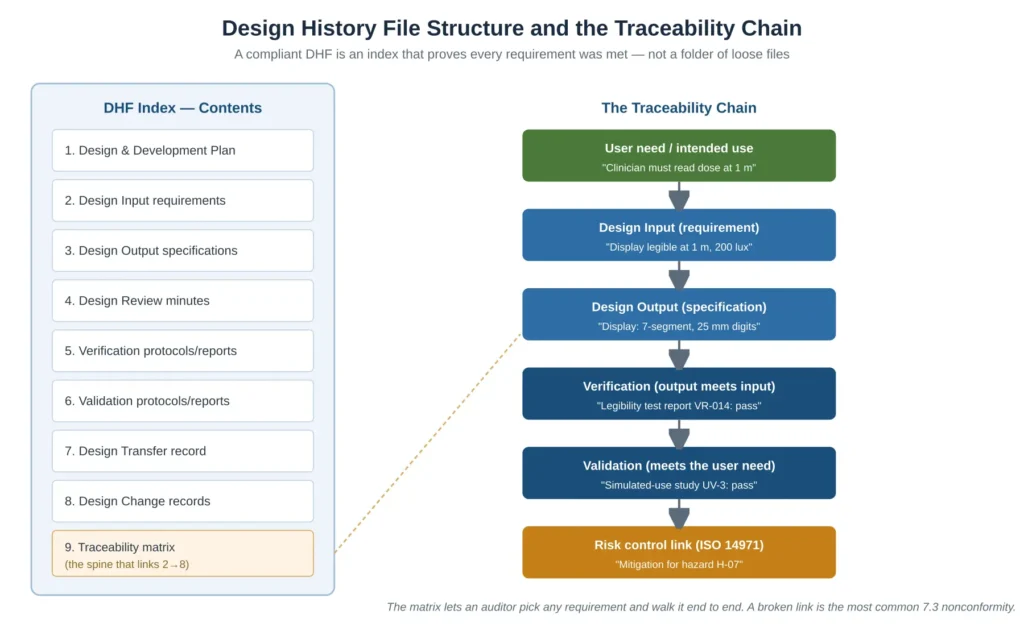
Figure 3 — Design History File structure and the requirement-to-validation traceability chain that auditors test
The practical discipline that prevents an incomplete DHF is to build the index from the start of the project, populate it continuously as each record is approved, and never treat DHF assembly as an end-of-project documentation exercise. A DHF assembled retrospectively almost always has gaps where intermediate decisions were made informally and never recorded — and those gaps cannot be back-filled honestly after the fact.
✦ PREMIUM BUNDLE · EU MDR COMPLETE
The complete EU MDR documentation set — technical, clinical and risk.
The Technical Documentation Kit (Annex II), the Clinical Documentation Kit (Articles 32, 61, 83-86) and the Risk Management Kit (ISO 14971 + IEC 62366-1) in one premium bundle. 22 templates, cross-referenced, with a single Master Index.
- ✓22 templates · pre-market + post-market documentation
- ✓Save €178 vs buying the three kits separately
Common Design Control and DHF Audit Findings
Across Notified Body and FDA-equivalent audits, the same design control deficiencies recur. Figure 4 sets out the six most common, colour-coded by typical severity, each paired with the corrective approach. Three of these — an incomplete DHF, broken traceability, and validation that is really only verification — routinely escalate to major or systemic nonconformities because each undermines the credibility of the entire design file rather than a single record.
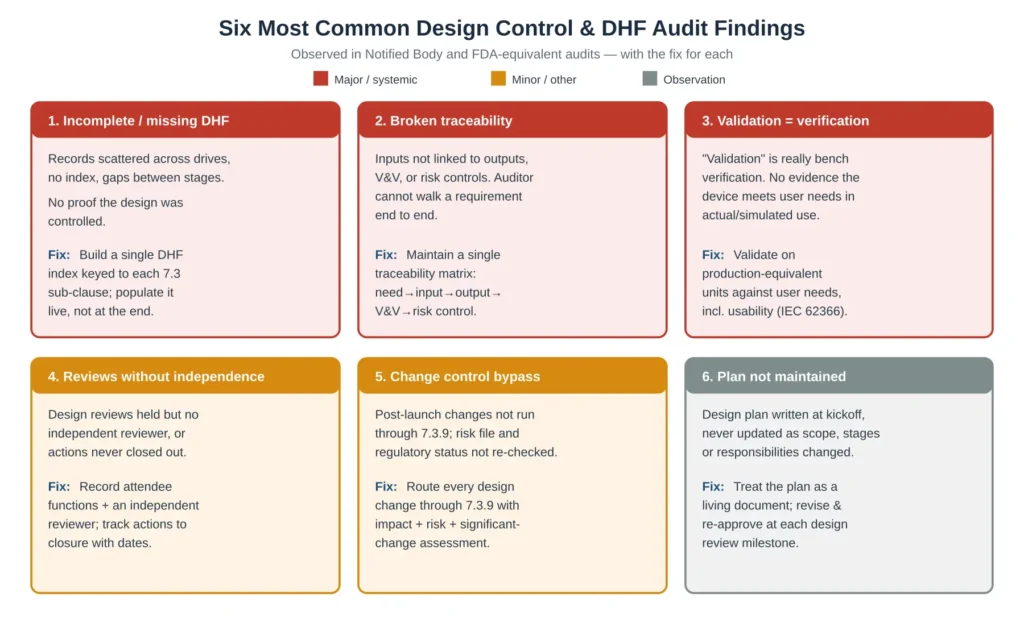
Figure 4 — The six most common design control and Design History File audit findings, severity-coded, with the fix for each
The pattern across all six is the same: design controls fail not because the activities were never done, but because they were done informally and the objective evidence linking them was never created or maintained. An auditor cannot give credit for work that is not demonstrable. The defence is structural — a file whose architecture forces completeness and traceability rather than relying on individual diligence. The same logic underpins audit readiness generally, as covered in our ISO 13485 internal audit checklist.
Frequently Asked Questions
What is the difference between a Design History File and a Device Master Record?
The Design History File is the record of how the design evolved — the plan, inputs, outputs, reviews, verification and validation evidence, transfer and change records. The Device Master Record (in ISO 13485 terms, the Medical Device File content) is the set of approved specifications and instructions used to actually manufacture the device. The DHF proves the design was controlled; the Medical Device File tells production how to build it. The design outputs are the bridge between them.
Are design controls required for Class I medical devices?
Under ISO 13485:2016 Clause 7.3, design and development controls apply regardless of device class, although the standard allows the extent of activities to be appropriate to the device. Historically the FDA exempted most Class I devices from design controls, but with the QMSR incorporating ISO 13485:2016 by reference from February 2026 the ISO structure now applies. The proportionality is in depth and rigour, not in whether the requirement applies at all.
How long must a Design History File be retained?
ISO 13485:2016 Clause 4.2.5 requires records to be retained for at least the lifetime of the medical device as defined by the organisation, but not less than two years from product release or as required by applicable regulatory requirements — whichever is longer. For long-lifetime implantable or capital equipment this can mean retention for well over a decade, so the DHF retention period should be defined explicitly and justified.
What is the difference between design verification and design validation?
Verification confirms that the design outputs meet the design inputs — it checks the device was built to specification. Validation confirms that the device meets the user’s needs and intended use under actual or simulated use conditions — it checks the specification was the right one. Verification can pass while validation fails if the inputs were wrong. Both are mandatory and their records must be distinct.
Does ISO 13485 require a specific Design History File format?
No. ISO 13485:2016 specifies the records that must exist and that they be maintained in a design and development file, but it does not mandate a template or tool. What it does require, in effect, is traceability: the file must allow any requirement to be traced through to its verification and validation. A simple indexed structure with a traceability matrix satisfies the standard; an elaborate tool with broken traceability does not.
When does a design change require regulatory notification?
ISO 13485 Clause 7.3.9 requires the impact of every change to be assessed, including its effect on product already delivered and on risk management. Whether a change additionally triggers regulatory notification depends on jurisdiction-specific significant-change rules under the EU MDR and FDA frameworks. The design change procedure should include an explicit step that routes every change through a significant-change assessment so that notification decisions are made deliberately and recorded, not made by omission.
Conclusions
The ISO 13485 design control requirements are not satisfied by performing design activities — they are satisfied by performing them in a controlled, traceable, recorded way that an auditor can independently verify. The practical takeaways are consistent: keep the design plan alive rather than fossilised; write inputs that are testable and trace every risk control into them; maintain bidirectional traceability between inputs and outputs; ensure design reviews are independent and their actions closed; never let validation collapse into verification; document design transfer rather than assuming manufacturability; route every change through risk and significant-change assessment; and above all build the Design History File continuously, as an index that forces completeness, rather than assembling it retrospectively when the gaps can no longer be filled honestly.
Get the structure right and Clause 7.3 stops being the highest-risk sampling path in your audit and becomes the part of the quality system that most clearly demonstrates the device is safe and effective. The ISO 13485 QMS Documentation Package on MD Regulatory includes the design and development procedure, design plan, input and output templates, design review minutes, verification and validation protocol templates, design transfer and design change records, and a ready-to-use Design History File index and traceability matrix — all aligned with ISO 13485:2016 Clause 7.3 requirements and immediately deployable in an existing ISO 13485 quality management system.
Related articles: ISO 13485 Complete Guide; ISO 13485 Internal Audit Checklist; ISO 13485 CAPA Procedure; ISO 14971 Risk Management; IEC 62304 Complete Guide.






One Comment
Comments are closed.