ISO 13485 Internal Audit Checklist: Clause-by-Clause Guide with Common Findings
Table of Contents
ISO 13485 internal audits are one of the most operationally critical — and most frequently misunderstood — requirements of ISO 13485:2016. When performed correctly, they are the most powerful tool a quality team has for identifying weaknesses before a Notified Body or FDA inspector does. When performed inadequately, they become a liability: a documented record that the organization looked at its own QMS and failed to find what regulators later found.
This guide covers everything you need to plan, conduct, document, and follow up on ISO 13485 internal audits effectively — including a complete clause-by-clause checklist, the most common audit findings, and practical guidance on how to structure your annual audit program.
AUDIT-READY KIT
Build your ISO 13485 QMS with confidence.
Built on 15+ years of audit experience — every SOP and template references the regulations auditors expect. Get to certification faster, with industry best practices baked in.
- ✓30 SOPs covering the full QMS scope
- ✓56 templates ready to customize
- ✓Aligned with EU MDR + FDA QMSR
What ISO 13485 Requires for Internal Audits
The internal audit requirement is contained in Clause 8.2.4 of ISO 13485:2016. The standard requires that organizations conduct internal audits at planned intervals to determine whether the QMS conforms to the requirements of ISO 13485 and to the QMS requirements established by the organization, and is effectively implemented and maintained.
This single clause carries significant operational weight. It requires:
A documented procedure covering audit responsibilities, planning requirements, conduct of audits, reporting, and follow-up. The procedure is mandatory — not optional.
A planned audit program — audits must be scheduled in advance, with frequency based on the status and importance of processes and the results of previous audits. An ad hoc or reactive approach does not satisfy the standard.
Auditor independence — personnel must not audit their own work. This is one of the most frequently violated requirements in small organizations where quality staff are also responsible for the processes being audited.
Records of audit results — including the audit plan, audit report, and evidence of follow-up on findings. Records must be retained per the organization’s documented retention requirements.
Timely corrective action — management responsible for the area being audited must take action without undue delay to eliminate detected nonconformities and their causes. Corrective actions must be verified for effectiveness.
How to Structure Your Annual Internal Audit Program
The annual audit program is the foundation of a compliant internal audit system. It is not a single audit — it is a planned series of audits that, taken together, cover the entire scope of the QMS within a defined period, typically one year.
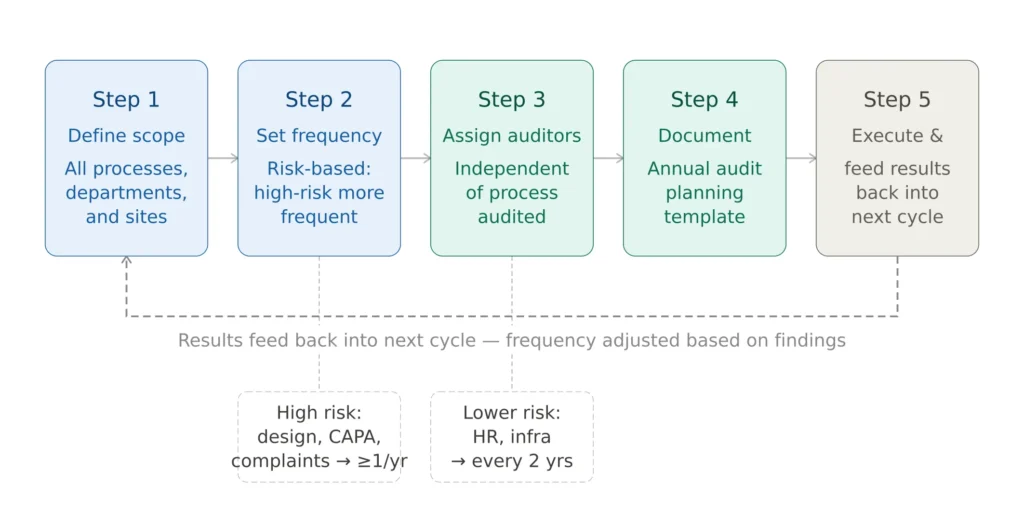
Figure 1 — ISO 13485 annual internal audit program structure
Step 1 — Define the scope of the program. The audit program must cover all processes, departments, and sites within the scope of your QMS. For manufacturers with a single site and a focused product portfolio this is relatively straightforward. For multi-site organizations or complex product portfolios, the program requires careful planning to ensure nothing is missed.
Step 2 — Determine audit frequency based on risk. ISO 13485 requires that audit frequency reflects the status and importance of the processes and the results of previous audits. Processes with previous nonconformities should be audited more frequently. High-risk processes — design and development, production, CAPA, complaint handling — typically warrant at least annual coverage. Lower-risk support processes such as HR and infrastructure may be audited less frequently if previous audits have been consistently satisfactory.
Step 3 — Assign auditors. Each audit must be conducted by personnel who are independent of the process being audited and who have the competence to assess it. Auditor competence must be documented — training records, lead auditor qualifications, or evidence of relevant experience.
Step 4 — Document the program. The annual audit program should be documented in an Annual Audit Planning Template that maps each QMS process against the planned audit schedule, assigned auditor, and audit scope. This document is a standard request item in Notified Body Stage 1 audits and FDA pre-inspection information requests.
Step 5 — Execute and feed back results. Results of each completed audit inform future audit frequency and focus — processes where findings were identified move to higher frequency in the next cycle.
Expand globally without rebuilding your QMS five times.
Single audit, five markets. Our MDSAP Kit gives you the documentation framework that works across all jurisdictions.
Explore the Kit →How to Plan and Conduct an Individual Internal Audit
Before the Audit — Preparation
Develop an audit plan. Each individual audit within the program requires its own audit plan covering: audit scope and objectives, audit criteria (which clauses or procedures apply), audit dates and location, auditor assignment, and the processes or departments to be audited. The audit plan should be communicated to the auditee in advance.
Review previous audit results. Before conducting the audit, the auditor should review findings from the previous audit of the same process — both to verify that previous corrective actions have been implemented and to identify areas requiring additional focus.
Prepare the checklist. The audit checklist is the auditor’s working document during the audit. It should be based on the relevant clauses of ISO 13485, the organization’s documented procedures, and any regulatory requirements applicable to the process being audited. A good checklist is a tool, not a script — it should prompt the auditor to investigate rather than simply confirm.
During the Audit — Conduct
Opening meeting. Begin with a brief opening meeting with the auditee to confirm the audit scope, objectives, and methodology, and to agree on logistics. This is not a formality — it sets the tone for the audit.
Evidence gathering. Internal audits are evidence-based assessments. The auditor collects objective evidence through three methods: interviews with personnel, observation of activities and conditions, and review of documents and records. No finding should be recorded without objective evidence to support it.
Sampling. It is not possible or necessary to review every record — auditors use sampling to assess the effectiveness of a process. The sample size should be sufficient to provide confidence in the conclusion and representative of the full range of activities within the process.
Document findings. During the audit, the auditor records findings as they are identified. Findings fall into three categories: nonconformities (failures to meet a stated requirement), observations or opportunities for improvement (potential weaknesses not yet constituting nonconformities), and positive findings (areas of particularly good practice).
Closing meeting. At the end of the audit, present findings to the auditee to confirm that findings are factually accurate and that the auditee understands what objective evidence supports each one.
After the Audit — Reporting and Follow-Up
Issue the audit report. The audit report must be issued promptly after the audit — within the timeframe defined in your procedure, typically five to ten working days. It must include: audit scope and objectives, audit criteria, auditor and auditee identification, dates, a summary of findings, and the overall audit conclusion.
Initiate corrective action. Each nonconformity must be addressed through the CAPA system. The responsible manager must define the immediate correction, root cause analysis, corrective action, and an implementation timeline. The auditor or quality manager must verify that the corrective action has been effectively implemented before closing the finding.
ISO 13485 Internal Audit Checklist — Clause by Clause
The following checklist covers the key requirements of ISO 13485:2016 organized by clause, designed as a working document during the audit.
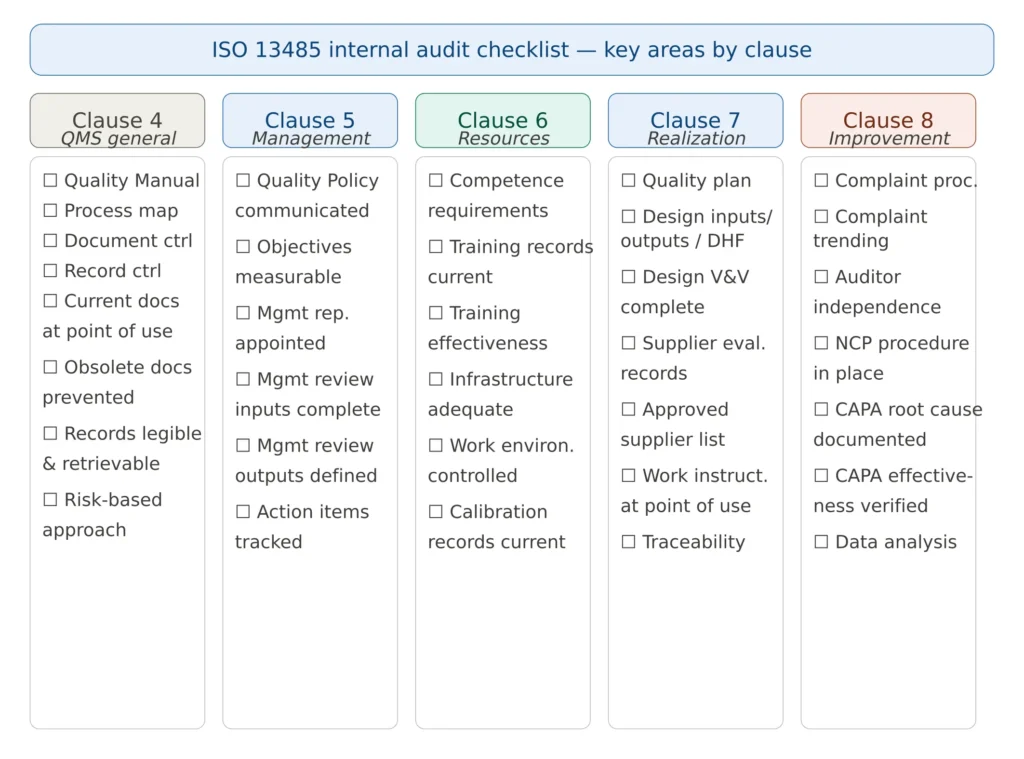
Figure 2 — ISO 13485 internal audit checklist structure by clause
Below is the full detailed checklist for use during audits, organized clause by clause.
Clause 4 — Quality Management System
- Is there a documented Quality Manual defining the scope of the QMS, including any exclusions and their justification?
- Are the processes of the QMS identified, sequenced, and their interactions defined?
- Are documented procedures in place for document control (4.2.3) and record control (4.2.4)?
- Are current versions of all applicable documents available at points of use?
- Are obsolete documents prevented from unintended use?
- Are records legible, identifiable, retrievable, and retained for the defined period?
- Is there evidence of a risk-based approach to process control?
Clause 5 — Management Responsibility
- Is there a documented Quality Policy that is communicated and understood by relevant personnel?
- Are Quality Objectives established, measurable, and monitored?
- Has a Management Representative been appointed with defined responsibilities?
- Is there evidence of management reviews conducted at planned intervals?
- Do management review records include all required inputs: audit results, customer feedback, process performance, CAPA status, regulatory changes, recommendations for improvement?
- Do management review outputs include decisions on resources, process improvements, and product improvements?
- Are action items from previous management reviews tracked to closure?
Clause 6 — Resource Management
- Are competence requirements defined for personnel performing work affecting product quality?
- Are training records maintained and current for all relevant personnel?
- Is there evidence that training effectiveness has been evaluated?
- Is infrastructure (buildings, equipment, IT systems) adequate and maintained?
- Are work environment conditions defined and controlled where applicable?
Clause 7 — Product Realization
7.1 — Planning: Is there a documented quality plan for each product or project? Are verification and validation activities planned and documented?
7.2 — Customer-related processes: Are product requirements — including regulatory requirements — identified and reviewed before acceptance? Is there a documented process for customer communication, including complaint handling?
7.3 — Design and Development: Is there a design and development plan for each device? Are design inputs documented and reviewed for adequacy? Are design outputs documented and traceable to design inputs? Have design reviews, verification, and validation been completed and documented? Is there a Design History File (DHF) for each device? Are design changes controlled through a documented change control process? Has design transfer to production been formally documented?
7.4 — Purchasing: Is there a documented procedure for supplier evaluation and selection? Is there an approved supplier list, maintained and current? Are purchasing documents adequate — specifying product requirements, quality requirements, and applicable regulatory requirements? Are suppliers monitored against defined criteria?
7.5 — Production and service provision: Are manufacturing processes performed under controlled conditions using documented work instructions? Are in-process inspections performed and recorded? Is product identification and traceability maintained throughout production?
7.6 — Control of monitoring and measuring equipment: Is all measurement equipment used to verify product conformity identified and calibrated? Are calibration records maintained, including the calibration standard used? Is the validity of previous measurements assessed when equipment is found out of calibration?
Clause 8 — Measurement, Analysis and Improvement
- Is there a documented procedure for complaint handling?
- Are complaints recorded, investigated, and trended?
- Are regulatory reporting obligations assessed for each complaint?
- Are internal audits planned and conducted per a documented procedure?
- Is auditor independence maintained?
- Are audit results reported to management?
- Is there a documented procedure for controlling nonconforming product?
- Is there a documented CAPA procedure covering root cause analysis, action implementation, and effectiveness verification?
- Are CAPAs opened for all nonconformities of significance?
- Is root cause analysis documented and appropriate to the significance of the nonconformity?
- Is effectiveness of corrective actions verified before CAPA closure?
Most Common ISO 13485 Internal Audit Findings

Figure 3 — Most common ISO 13485 internal audit findings
Design and development controls remain the single most frequently cited area of nonconformity in ISO 13485 audits globally — incomplete design inputs, missing verification or validation records, undocumented design changes, or absence of a formal design transfer procedure.
CAPA effectiveness is a persistent finding: CAPAs opened and closed without adequate root cause analysis, or without documented evidence that the effectiveness of the corrective action was verified. Closing a CAPA on time is not the same as closing it effectively.
Supplier management weaknesses — suppliers approved without documented evaluation, approved supplier lists not maintained or not linked to purchasing documents, or critical suppliers not subjected to periodic performance monitoring.
Auditor independence — in small organizations, the quality manager audits processes they are directly responsible for. This is a structural nonconformity that cannot be addressed by adding caveats to the audit report.
Management review inputs — reviews conducted but without evidence that all required inputs were reviewed, or with inputs that are superficial and not data-driven. Notified Bodies look for evidence of genuine top management engagement, not a compliance exercise.
Calibration — measurement equipment used in production or inspection without current calibration, calibration records that do not identify the reference standard used, or no process for assessing the impact of out-of-tolerance equipment on previously released product.
Record retention — records not retained for the required period, records not identifiable or retrievable, or no documented retention policy aligned with regulatory requirements.
Training effectiveness — training records demonstrate attendance, but there is no evidence that training effectiveness was evaluated or that personnel can apply the requirements in practice.
✦ PREMIUM BUNDLE
The ultimate global QMS documentation bundle.
Combine ISO 13485 + all 5 MDSAP markets in one premium package. Deduplicated structure means you customize each document once — not twice. Save €199 vs buying the kits separately.
- ✓41 SOPs covering both ISO 13485 and MDSAP
- ✓70+ templates with deduplicated structure
- ✓Save €199 vs buying separately
How to Write a Nonconformity Correctly
A well-written nonconformity statement is essential for effective corrective action. It must contain three elements: the objective evidence observed, the requirement that is not being met, and the clause or procedure reference.
A poorly written nonconformity: “Supplier management is inadequate.”
A correctly written nonconformity: “During the audit of the purchasing process, it was observed that Supplier X (reference PO-2025-0342) is listed on the Approved Supplier List but no supplier evaluation records were found. This does not meet the requirements of ISO 13485:2016 Clause 7.4.1, which requires that suppliers be evaluated and selected based on their ability to meet specified requirements, and that records of evaluations be maintained.”
The difference matters — a vague finding produces a vague corrective action. A specific, evidence-based finding enables meaningful root cause analysis and targeted corrective action.
Internal Audit Records — What You Must Retain
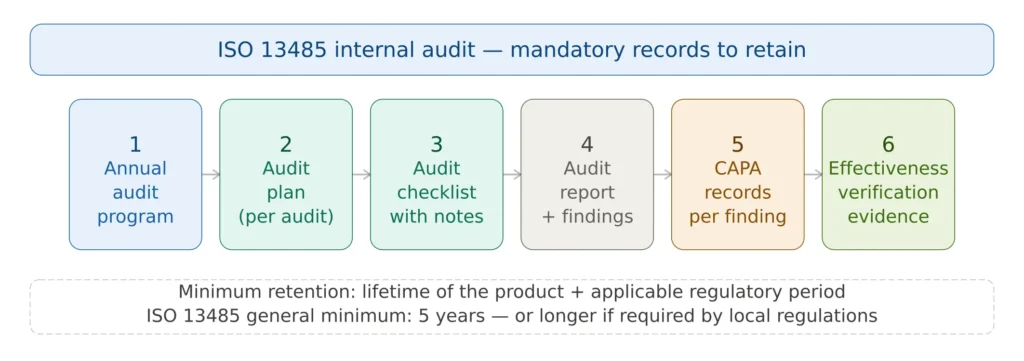
Figure 4 — ISO 13485 internal audit record hierarchy
ISO 13485 requires that records of audit results be maintained. At a minimum, the following six records should be retained for each audit: the annual audit program, the audit plan issued in advance for each individual audit, the completed audit checklist with evidence notes, the formal audit report including all findings, CAPA records linked to each nonconformity, and effectiveness verification records demonstrating that corrective actions worked.
These records should be retained for a minimum period defined by your document control procedure — typically the lifetime of the product plus the retention period required by applicable regulations, or a minimum of five years for most ISO 13485-certified organizations. Under FDA QMSR, internal audit records are now also subject to FDA inspection.
Frequently Asked Questions
How often must internal audits be conducted under ISO 13485? ISO 13485 requires audits at planned intervals but does not specify a minimum frequency. Industry practice and Notified Body expectations typically require full QMS coverage at least once per year, with higher-risk or previously nonconforming processes audited more frequently.
Can the same person plan and conduct the internal audit? Yes — the same person can plan and conduct an audit, provided they are independent of the process being audited. What is not permitted is auditing your own work.
Can we use an external consultant to conduct our internal audits? Yes. Using a qualified external auditor is a legitimate and frequently used approach, particularly for small organizations that lack internal auditor resources. The external auditor must have documented competence in ISO 13485 and must be given access to all relevant processes and records.
What happens if we find a major nonconformity during an internal audit? A major nonconformity found during an internal audit must be addressed through your CAPA system with priority. It should be reported to management immediately and addressed before your next certification or surveillance audit. Finding it yourself — and correcting it — is significantly better than a Notified Body finding it first.
Do internal audit findings need to be reported to the Notified Body? Individual internal audit findings do not need to be proactively reported. However, your internal audit records — including findings and corrective actions — are reviewed by the Notified Body during surveillance audits and must be available for inspection. Under FDA QMSR, internal audit records are now also subject to FDA inspection.
Conclusions
A well-executed internal audit program is not a compliance burden — it is the most cost-effective quality investment a medical device manufacturer can make. Every nonconformity identified and corrected internally costs a fraction of the time, resources, and reputational impact of the same finding identified by a Notified Body, FDA inspector, or — worst of all — through a field event.
The organizations that consistently achieve clean audit outcomes are not those with the most sophisticated QMS on paper. They are the ones that audit rigorously, write honest findings, investigate root causes thoroughly, and verify that their corrective actions actually work.
If you are building or upgrading your internal audit system, the right documentation framework is the essential starting point. The Audit Management SOP package available on MD Regulatory includes a complete internal audit procedure, annual audit planning template, audit plan template, and audit report template — all written to reflect current Notified Body expectations and immediately usable in your QMS.
This article is part of the MD Regulatory ISO 13485 series. Related articles: ISO 13485 Complete Guide, ISO 13485 CAPA Procedure, ISO 13485 Supplier Qualification, ISO 13485 Management Review.




2 Comments
Comments are closed.