EU MDR Post-Market Surveillance : PMS Plan, PSUR and PMCF Requirements
Table of Contents
EU MDR post-market surveillance is one of the most operationally demanding requirements of Regulation (EU) 2017/745 — and the area where the gap between what the MDR demands and what manufacturers actually have in place is widest. Unlike the previous Medical Devices Directive, which treated PMS as a general obligation, the MDR establishes PMS as a structured, proactive, and continuous process with explicit documentation requirements, defined deliverables, and direct links to clinical evaluation and risk management.
For manufacturers that have achieved CE marking under EU MDR or are in the process of transitioning, the post-market surveillance system is not a one-time deliverable — it is an ongoing operational commitment that must be resourced, maintained, and continuously fed with real-world data throughout the entire commercial lifetime of the device.
This guide covers the complete EU MDR post-market surveillance framework: the PMS plan structure, the Periodic Safety Update Report (PSUR) and Post-Market Surveillance Report (PMSR) requirements by device class, Post-Market Clinical Follow-Up (PMCF) obligations, and how to build a surveillance system that actually works in practice. For context on how PMS integrates with the broader technical documentation requirements, see our EU MDR technical documentation guide.
✦ EU MDR CLINICAL DOCUMENTATION KIT
Clinical evaluation, PMS, PMCF and SSCP — covered end-to-end.
8 Word templates for the full clinical lifecycle under EU MDR — CEP, CER, CDP, PMS Plan, PSUR, PMCF Plan and Evaluation Report, plus SSCP for Class III and implantables. Aligned with MDCG 2020-5/6/7/8/13 and 2022-21.
- ✓8 Word templates · pre-market + post-market
- ✓MDCG-aligned · ready for Notified Body submission
What EU MDR Requires for Post-Market Surveillance
EU MDR 2017/745 addresses post-market surveillance across several articles and annexes, creating an interconnected system of obligations:
Article 83 establishes the general requirement: every manufacturer must plan, establish, document, implement, maintain, and update a post-market surveillance system for each device. The system must be an integral part of the manufacturer’s ISO 13485 quality management system.
Article 84 requires that the PMS plan be part of the technical documentation (Annex III). It must proactively collect and review experience gained in the post-production phase.
Article 85 requires manufacturers of Class I devices to produce a Post-Market Surveillance Report (PMSR).
Article 86 requires manufacturers of Class IIa, IIb, and III devices to produce a Periodic Safety Update Report (PSUR), with update frequencies that vary by device class.
Article 87-92 define the vigilance system — serious incident reporting, field safety corrective actions, and trend reporting — which operates in parallel to, but is distinct from, the PMS system.
Annex XIV Part B defines PMCF requirements — the clinical component of post-market surveillance.
Annex III defines the technical documentation content for PMS, including the specific elements that must be covered in the PMS plan.
The absence of an effective PMS system may lead to certification delays, product withdrawals from the market, or even administrative sanctions. Many manufacturers face difficulties in implementing a surveillance system that not only meets formal regulatory requirements but also effectively detects risks and supports quality improvement.
The PMS System — How It Works
EU MDR post-market surveillance is not a single document — it is a system of interconnected processes, data sources, analysis activities, and outputs that operate continuously throughout the device lifecycle.
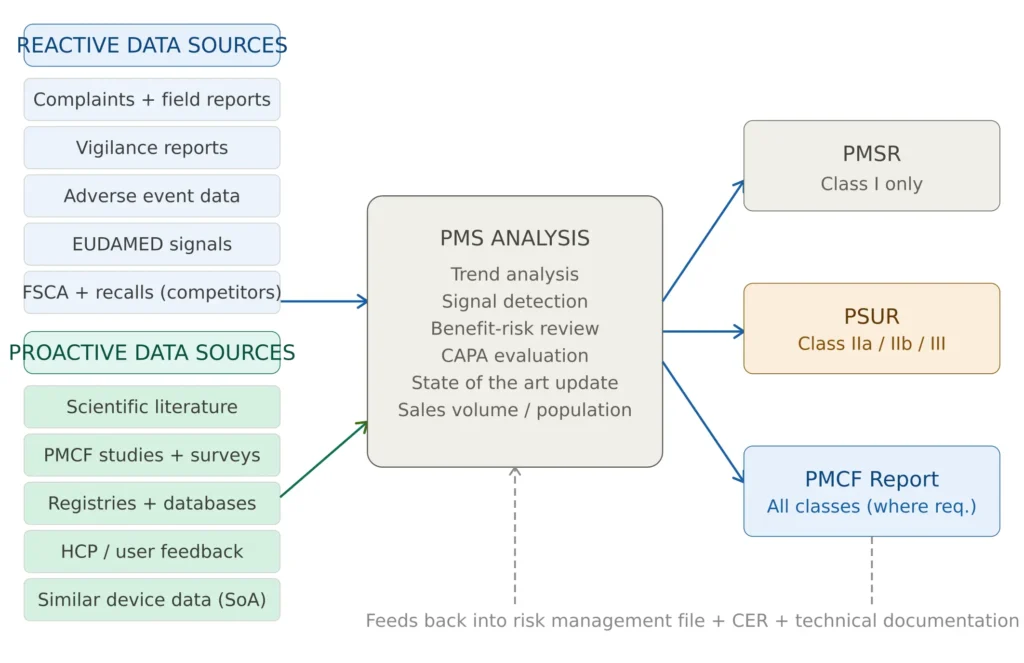
Figure 1 — EU MDR post-market surveillance system overview
The Post-Market Surveillance Plan (PMS Plan)
The PMS plan is the foundational document of the entire surveillance system. The post-market surveillance plans should be established before placing the device on the market and be updated throughout the lifetime of the device as needed. It is part of the technical documentation required under Annex III of EU MDR and must be device-specific — a generic template applied across product lines is not acceptable.
Mandatory content of the PMS Plan under Annex III:
Data sources to be monitored — both reactive and proactive. Reactive sources include complaint data, vigilance reports from EUDAMED, adverse events reported by users and healthcare professionals, and field safety corrective actions from competitors. Proactive sources include systematic literature reviews, clinical registries, post-market clinical investigations, post-market clinical follow-up surveys, and real-world evidence databases.
Methods for collecting and analyzing data — the PMS plan must specify how data from each source will be retrieved, how frequently, and how it will be analyzed. Statistical methods for trend detection must be defined, including the thresholds that would trigger a signal investigation or CAPA.
Performance and safety indicators — specific, measurable KPIs against which device performance will be assessed in the field. These must be consistent with the performance claims and safety thresholds established in the clinical evaluation and risk management file.
Reference to PMCF plan — both the EU MDR and IVDR require that the PMCF be addressed in the manufacturer’s post-market surveillance plan. In brief, the PMCF plan must be part of the corresponding PMS plan.
Link to the vigilance system — while vigilance reporting and PMS are distinct processes, the PMS plan must describe how vigilance data feeds into the PMS analysis. It is a common misconception to consider that the PMS thresholds and indicators are the same as the triggers for vigilance trend reporting. Although there is some overlap, they are not exactly the same.
Update triggers — conditions under which the PMS plan will be reviewed and updated, including new safety signals, significant changes to the device, changes in clinical practice or state of the art, and outputs from the PSUR or PMCF report.
PMSR vs PSUR — Requirements by Device Class
One of the most common points of confusion in EU MDR post-market surveillance is which report is required for which device class and at what frequency. The following clarifies the requirements definitively.
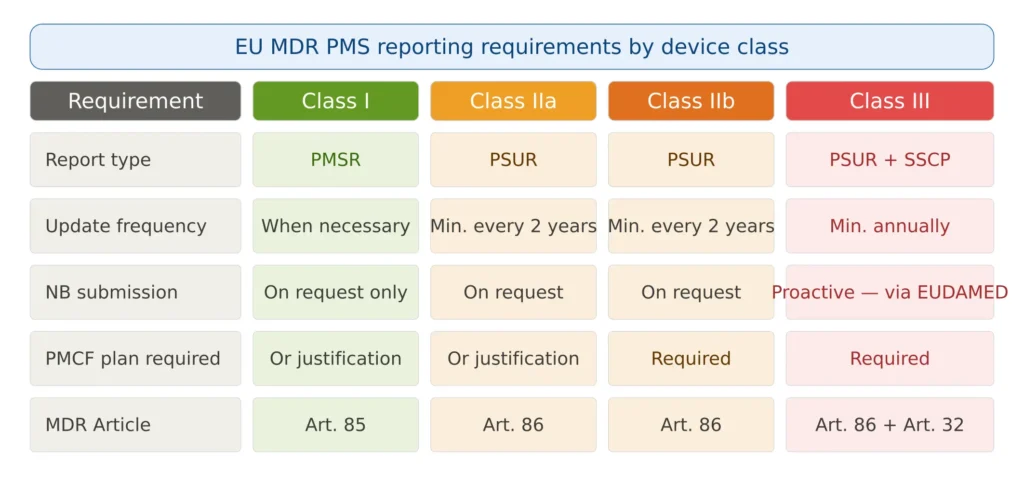
Figure 2 — PMS reporting requirements by device class
The Post-Market Surveillance Report (PMSR) — Class I Devices
The PMSR is required for Class I device manufacturers under Article 85. It is simpler than the PSUR but still requires substantive content — it is not a brief summary memo.
The PMSR must include: a summary of the results and conclusions from analyzing the PMS data collected under the PMS plan; a rationale and description of any preventive and corrective actions taken as a result of PMS findings; and confirmation that the benefit-risk profile of the device remains acceptable.
A Post-market Surveillance Report is only required for manufacturers of Class I medical devices. The PMSR must be available to competent authorities upon request. Unlike the PSUR, the PMSR does not need to be proactively submitted to a Notified Body — but it must be maintained and available for inspection.
The Periodic Safety Update Report (PSUR) — Class IIa, IIb, and III
The PSUR is the more comprehensive PMS output required for Class IIa, IIb, and III devices. Article 86 of the EU MDR details that the PSUR must include the conclusions of the benefit-risk determination, the main findings of the Post Market Clinical Follow-up (PMCF), the sales volume of the device, and an estimate of the size and other characteristics of the population using the device including frequency of use when applicable.
The MDCG 2022-21 guidance provides the most detailed template for PSURs currently available. Key sections that every PSUR must address include:
Device identification and scope — the device name, basic UDI-DI, device description, intended purpose, and the period covered by the PSUR.
PMS data summary — a structured review of all data collected from the PMS plan data sources during the reporting period, including complaint volumes, serious incident rates, field safety corrective actions, and literature findings.
Benefit-risk determination — an updated benefit-risk analysis based on the PMS data, confirming that the benefit-risk balance remains acceptable or documenting the actions taken when it does not. This connects directly to the benefit-risk analysis maintained in the technical documentation.
Clinical evaluation update — a summary of how PMS data has been used to update the clinical evaluation, with conclusions on whether the clinical evidence remains sufficient to support the intended purpose. The PSUR effectively triggers the periodic update of the Clinical Evaluation Report (CER).
PMCF findings — the main findings from PMCF activities conducted during the reporting period, including any new data on long-term safety or performance, rare complications, or off-label use.
Sales volume and population data — the number of devices sold or distributed during the period and an estimate of the patient/user population, including frequency of use.
Corrective and preventive actions — all CAPAs initiated as a result of PMS findings during the period, with implementation status and effectiveness verification.
PSUR update frequency:
Manufacturers of Class IIa medical devices must update the PSUR when necessary, but at least every two years. Manufacturers of Class IIb and Class III medical devices must update the PSUR at least annually.
For Class III and implantable Class IIb devices, the PSUR must be made available to the Notified Body via EUDAMED and is subject to review as part of ongoing surveillance obligations. PSURs for Class III and implantable devices must be submitted to the Notified Body via EUDAMED and will be subject to review as part of surveillance obligations.
For Class IIa and non-implantable Class IIb devices, the PSUR must be maintained and made available to the Notified Body and competent authority on request.
✦ PREMIUM BUNDLE · EU MDR COMPLETE
The complete EU MDR documentation set — technical, clinical and risk.
The Technical Documentation Kit (Annex II), the Clinical Documentation Kit (Articles 32, 61, 83-86) and the Risk Management Kit (ISO 14971 + IEC 62366-1) in one premium bundle. 22 templates, cross-referenced, with a single Master Index.
- ✓22 templates · pre-market + post-market documentation
- ✓Save €178 vs buying the three kits separately
Post-Market Clinical Follow-Up (PMCF)
PMCF is the clinical component of post-market surveillance — and the element that most commonly generates major findings in Notified Body surveillance audits. PMCF is a continuous process in which the manufacturer continuously and proactively collects and evaluates clinical data. PMCF data can support the clinical evidence of a device and may reduce the need to conduct additional clinical investigations.
PMCF is defined in Annex XIV Part B of EU MDR and must be addressed in the PMS plan for all device classes. For Class IIb and III devices, a PMCF plan and PMCF report are mandatory. For Class I and IIa devices, a PMCF plan is required or a documented justification for why PMCF is not applicable.
The PMCF Plan
The PMCF plan must define:
Objectives — specific clinical questions that PMCF is designed to answer. These must address data gaps identified in the clinical evaluation — long-term safety outcomes, rare adverse events, performance in specific patient subgroups, or real-world performance versus controlled study conditions. Generic objectives (“to confirm device safety and performance”) without specific measurable endpoints are consistently flagged as deficient by Notified Bodies.
Methods — the specific data collection activities planned, including general methods applicable to any device (systematic literature surveillance, clinician/user surveys, analysis of complaint and registry data) and specific methods appropriate to the device risk class (post-market clinical investigations, observational studies, registry participation).
Timelines — when each PMCF activity will start, how long it will run, and when results will be analyzed and reported.
Acceptance criteria — what results would confirm that the clinical evidence remains adequate, and what results would trigger further action.
Justification where PMCF is claimed not applicable — if a manufacturer claims that no PMCF activities are required (for example because the clinical evidence from pre-market studies is comprehensive and the device has a long established history), this justification must be detailed, specific, and scientifically defensible. The absence of an effective PMCF plan is one of the most frequently cited major findings in Notified Body surveillance audits — particularly generic plans that do not define specific data collection methods, timelines, or analysis criteria relevant to the specific device.
The PMCF Report
The PMCF report summarizes the results of all PMCF activities conducted during the reporting period, evaluates whether the objectives defined in the PMCF plan were achieved, draws clinical conclusions from the data, and defines what further PMCF activities are needed.
The PMCF report must include analysis of PMCF data, conclusions on continued safety and performance, and any updates needed to the clinical evaluation, risk management, or benefit-risk determination.
The PMCF report feeds directly into three other documents: the PSUR (where the main PMCF findings are summarized), the Clinical Evaluation Report (which must be updated based on PMCF data), and the ISO 14971 risk management file(where PMCF findings that identify new hazards or change risk estimates must be incorporated).

Figure 3 — PMCF plan structure and required elements
The Summary of Safety and Clinical Performance (SSCP)
For Class III and implantable devices, EU MDR requires an additional public-facing document: the Summary of Safety and Clinical Performance (SSCP). The SSCP is validated by the Notified Body and published on EUDAMED — making it publicly accessible to patients, healthcare professionals, and other stakeholders.
The SSCP must be written in plain language understandable to non-specialists, and must cover: device description and intended purpose; indications, contraindications, and target patient population; summary of clinical evidence; information on any residual risks and undesirable side effects; recommendations for follow-up; suggested profile of the user; and reference to the applicable standards and harmonized standards.
The SSCP must be updated as part of the PSUR process — whenever new PMS data changes the benefit-risk determination or clinical conclusions.
How PMS Connects to the Rest of the Technical Documentation
Post-market surveillance is not a standalone system — it is the continuous feedback loop that keeps the entire EU MDR technical documentation current and compliant throughout the device lifecycle.
Risk management file: PMS findings that identify new hazards, change risk probability estimates, or reveal that existing risk controls are insufficient must be fed back into the ISO 14971 risk management file. The risk management file must be updated to reflect post-market findings, and the overall residual risk re-evaluated.
Clinical Evaluation Report: PMS data including PMCF findings must be used to update the clinical evaluation throughout the device lifecycle. The CER must reflect the current state of clinical evidence — not just the pre-market data. The PSUR typically triggers a review of the CER to assess whether an update is required.
ISO 13485 CAPA system: PMS signals that indicate potential safety issues or performance degradation must be processed through the CAPA procedure. The PMS system does not replace the CAPA system — it feeds into it.
EUDAMED: For Class III and implantable Class IIb devices, PMS outputs — particularly the PSUR and SSCP — must be uploaded to EUDAMED. The EUDAMED vigilance module also generates data that feeds back into the PMS system.
Vigilance system: While vigilance reporting (Article 87-89) and PMS are distinct processes, they are deeply interconnected. Serious incidents and field safety corrective actions identified through the vigilance system become data inputs to the PMS analysis. Conversely, PMS trend analysis may identify signals that require vigilance reporting even when individual incidents have not crossed the reporting threshold.
Most Common PMS Audit Findings

Figure 4 — Most common EU MDR PMS audit findings
Generic PMS plan — the most common finding. The PMS plan references data sources but does not define specific performance indicators, analysis thresholds, or acceptance criteria for the specific device. A compliant PMS plan must define what constitutes a signal for this device — a complaint rate threshold, an adverse event frequency, a literature finding — not just list generic data source categories.
Generic PMCF plan — closely related. PMCF plans that state “literature will be reviewed annually” without defining specific search terms, databases, inclusion/exclusion criteria, or acceptance thresholds are consistently rejected. The PMCF plan must be written as if it were a clinical study protocol — specific enough that an independent reviewer could execute it.
PSUR not updated on schedule — a process management failure. The annual or biennial PSUR update cycle must be embedded in the QMS calendar with defined owners, deadlines, and escalation paths. Missing a PSUR update cycle is a direct regulatory non-compliance.
PMS disconnected from risk management — PMS findings are documented in the PSUR but do not trigger updates to the risk management file or the clinical evaluation. The feedback loop from PMS to risk management is mandatory under ISO 14971 Clause 10 and must be documented with evidence.
No documented data analysis — data is collected from complaint systems, literature databases, and EUDAMED but there is no documented record of the analysis performed. Data analysis outputs — trend graphs, signal assessment records, literature appraisal logs — must be retained as objective evidence.
SSCP not synchronized with PSUR — for Class III and implantable devices, the SSCP on EUDAMED is not updated to reflect new clinical conclusions from the most recent PSUR. The SSCP must be reviewed as part of every PSUR cycle.
✦ EU MDR TECHNICAL DOCUMENTATION KIT
The complete Annex II technical file in one coordinated set.
8 Word templates covering the full EU MDR Annex II structure, plus Annex III (PMS) and Annex IV (Declaration of Conformity). Built around the latest MDCG guidance and Team-NB position papers. Suitable for all MDR classes (I, IIa, IIb, III) and custom-made devices.
- ✓8 templates · Annex II + III + IV
- ✓Team-NB position papers integrated as inline notes
Practical Guide — Building a Compliant PMS System
Integrate PMS into the QMS from day one. PMS is not a post-launch add-on — it must be planned before market placement. The PMS plan should be drafted as part of the technical documentation preparation, with data sources, KPIs, and PMCF activities defined before the device enters clinical use. This connects directly to ISO 13485 Clause 8.2 requirements for feedback and data analysis.
Define device-specific KPIs with measurable thresholds. For each performance or safety claim in the intended purpose, define a quantitative or semi-quantitative indicator. For example: complaint rate per 1,000 devices per year, percentage of devices requiring field service within 24 months, adverse event frequency for the primary risk, literature publication rate on device-related complications. These KPIs anchor the analysis and make signal detection objective.
Plan PMCF as a clinical research activity, not as a documentation exercise. PMCF studies — whether surveys, registry participation, or prospective observational studies — require ethical approval where applicable, patient informed consent, and appropriate statistical power to generate meaningful conclusions. A PMCF plan that proposes a 10-patient survey to address a safety gap in a 50,000-patient device population will not satisfy a Notified Body reviewer.
Establish formal data governance for literature surveillance. Literature surveillance is the most universally applicable PMCF method — but it must be conducted systematically. Define the databases to be searched (PubMed, Embase, Cochrane), the search terms, the inclusion and exclusion criteria, the frequency, and who is responsible. Document each search cycle with the date, results, and appraisal conclusion.
Automate PSUR cycle reminders in the QMS. The PSUR update frequency — annual for Class IIb and III, biennial for Class IIa — must be embedded in your document management system with automated reminders and defined responsibility. A missed PSUR is an immediate compliance gap with no grace period.
Use PSUR findings as active inputs to CER updates. Every PSUR should include an explicit statement about whether the clinical evaluation requires updating based on PMS findings. If it does, the CER update should be initiated within a defined timeframe and the updated CER should reference the specific PMS data that triggered the review.
Frequently Asked Questions
What is the difference between PMS and vigilance reporting? PMS and vigilance are complementary but distinct systems. PMS is a proactive, continuous process of collecting and analyzing post-market data to confirm device safety and performance — it operates at the system level and produces periodic outputs (PSUR, PMSR). Vigilance reporting is a reactive, event-driven process that requires notification to competent authorities when specific serious incidents or malfunctions occur — it operates at the incident level with defined reporting timelines (15 days for serious incidents). It is a common misconception to consider that the PMS thresholds and indicators are the same as the triggers for vigilance trend reporting. Although there is some overlap, they are not exactly the same.
Is PMCF mandatory for all medical devices? PMCF or a documented justification that PMCF is not applicable is required for all device classes. For Class I and IIa devices, a justified “no PMCF” decision may be acceptable if the clinical evidence is comprehensive and the device has an established safety and performance history. For Class IIb and III devices, PMCF is practically always required — justifications for no PMCF for these classes are extremely difficult to sustain under Notified Body scrutiny.
When must the PSUR be submitted to the Notified Body? For Class IIa and non-implantable Class IIb devices, the PSUR must be maintained and made available to the Notified Body and competent authority on request — there is no proactive submission requirement. For Class III and implantable Class IIb devices, the PSUR must be submitted to the Notified Body via EUDAMED and will be reviewed as part of the surveillance audit cycle.
Can the PMSR and PSUR reference the same PMS plan? Yes — all devices within a device family may share a PMS plan if they have the same intended purpose and similar risk profiles, with device-specific elements documented as appendices or supplements. However, each device must have its own PMSR or PSUR — aggregate reports covering multiple devices are not permitted.
How do EU MDR transition deadlines affect PMS obligations? Manufacturers of legacy devices transitioning under EU MDR transition deadlines must establish MDR-compliant PMS systems as part of their transition. The PMS plan and associated documentation must be ready before the MDR certificate is issued — not built after market placement. For devices transitioning under Regulation 2023/607, the PMS compliance clock is tied to the MDR certificate date, not the original MDD placement date.
Conclusions
EU MDR post-market surveillance represents the most fundamental shift in how medical device compliance is conceived in Europe — from a pre-market approval exercise to a continuous lifecycle commitment. PMS data including PMCF findings must be used to update the clinical evaluation throughout the device lifecycle, and the risk management file must reflect post-market findings continuously.
The manufacturers who consistently achieve clean Notified Body surveillance audit outcomes are not those with the most elaborate PMS software or the most comprehensive procedures. They are the ones who treat PMS as an active intelligence system — using complaint trends, PMCF data, and literature findings to genuinely understand how their device performs in the real world, and feeding those insights back into risk management, clinical evaluation, and product improvement decisions.
For manufacturers building or upgrading their PMS system, the right documentation framework is the essential starting point. The EU MDR PMS Documentation Package on MD Regulatory includes a PMS plan template aligned with Annex III, a PSUR template following MDCG 2022-21 guidance, a PMCF plan template with Annex XIV Part B compliance, and a PMSR template for Class I devices — all immediately deployable in an ISO 13485 quality management system.
This article is part of the MD Regulatory EU MDR series. Related articles: EU MDR Technical Documentation Requirements · EU MDR Transition Deadlines 2026/2027 · Benefit-Risk Analysis under EU MDR · ISO 14971 Risk Management · ISO 13485 CAPA Procedure





One Comment
Comments are closed.