IEC 62304: The Complete Guide to the Medical Device Software Standard
Table of Contents
Introduction
IEC 62304:2006 is the internationally recognized standard providing a framework for the development, testing, and maintenance of software used in — or as — medical devices. With the rapid growth of software-based technologies in the medical field (including Software as a Medical Device, SaMD), understanding and correctly applying this standard has become essential for manufacturers, software developers, and regulatory affairs professionals alike.
This article is the first in a series dedicated to IEC 62304. It covers the following foundational topics:
- Software Safety Classification
- How to apply the Software Safety Classification
- Software Development Plan
- Software Architecture
Future posts in this series will also address topics related to medical device cybersecurity, including cybersecurity risk assessment, threat modeling, patch management, cybersecurity testing, and vulnerability disclosure.
What Is IEC 62304?
IEC 62304:2006 — and its amendment IEC 62304:2006/AMD 1:2015 — defines the life cycle requirements for medical device software. It is a process-oriented standard: rather than dictating specific technical solutions, it establishes the activities, tasks, and documentation that must be in place throughout the entire software life cycle, from planning through maintenance.
The standard applies to:
- Software embedded in a medical device (e.g., firmware controlling an infusion pump)
- Software that is itself a medical device (e.g., a clinical decision support application)
- SOUP (Software of Unknown Provenance) — third-party or off-the-shelf software components integrated into a medical device
IEC 62304 is typically applied in conjunction with ISO 14971 (risk management for medical devices) and IEC 62366 (usability engineering), forming the core triad of software-related standards in the medical device regulatory landscape.
From standard to submission-ready file. Understanding IEC 62304 is step one; producing the documents is where audits are won. Our IEC 62304 Software Documentation Kit translates every clause covered in this guide into twelve clause-referenced, fully editable templates — development plan to release record, cybersecurity included, scalable to Class A, B or C.
✦ PREMIUM BUNDLE · SW DOCUMENTATION KIT GOLD
Software lifecycle, AI/ML and cybersecurity — the complete file for AI-enabled devices.
The full IEC 62304 lifecycle set, the complete AI/ML module built on the IMDRF GMLP principles (including the PCCP and the AI-augmented CEP/CER), and the cybersecurity documentation — 23 cross-referenced templates in three coordinated modules, with a Master Index & Conformity Matrix.
- ✓23 templates · 19 Word + 4 Excel · three coordinated modules
- ✓Save €199 vs buying the SW Kit and AI/ML Kit separately
Software Safety Classification Under IEC 62304
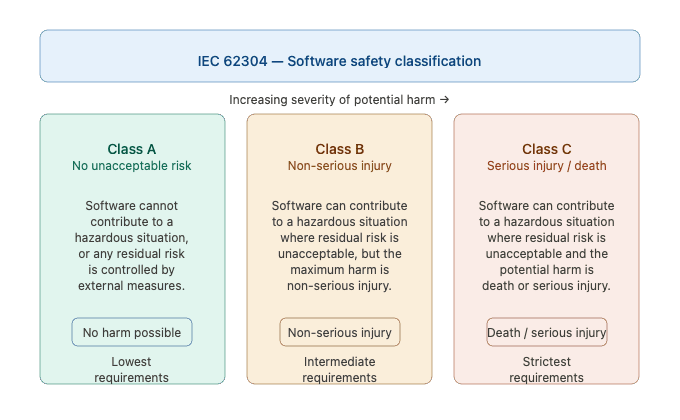
One of the most critical — and often misunderstood — aspects of IEC 62304 is the software safety classification system. The standard defines three classes, based on the severity of harm that a software failure could cause to patients, users, or third parties.
Class A — No Injury Risk
The software system cannot contribute to a hazardous situation, or it can contribute to one that does not result in unacceptable risk after the implementation of risk control measures external to the software itself.
Example: A software component that displays administrative data unrelated to patient treatment.
Class B — Risk of Non-Serious Injury
The software system can contribute to a hazardous situation that results in unacceptable risk, even after considering external risk control measures — but the maximum possible harm is limited to non-serious injury.
Example: Software that controls a low-risk therapy where a malfunction could cause discomfort but not permanent damage.
Class C — Risk of Serious Injury or Death
The software system can contribute to a hazardous situation resulting in unacceptable risk, where the potential harm is death or serious injury.
Example: Software managing drug dosage in an automated infusion system.
Key principle: The higher the safety class, the more rigorous the development, verification, and documentation requirements imposed by the standard.
How to Apply the Software Safety Classification
Correctly assigning a safety class is not always straightforward. A structured, requirement-based approach is one of the most reliable methods.
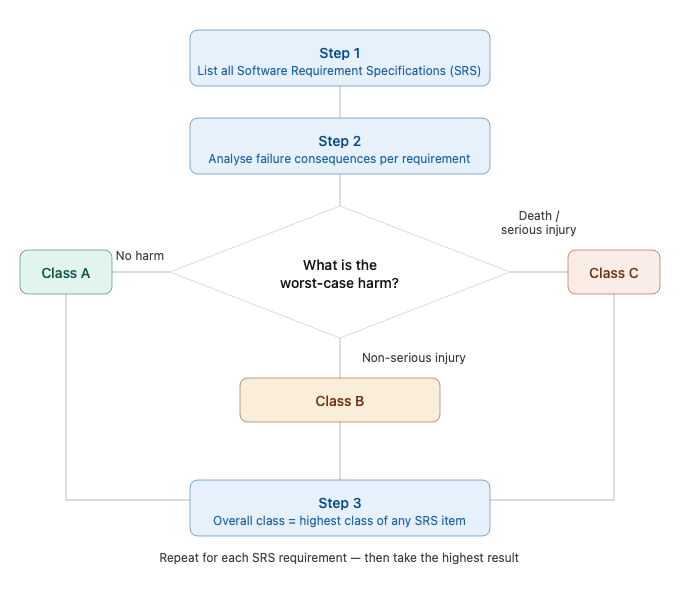
Here is a practical step-by-step process:
- List all Software Requirement Specifications (SRS). Document every functional and non-functional requirement of the software system.
- Perform a failure analysis for each requirement. For each requirement, ask: What happens if this requirement is not met or fails? Evaluate the potential consequences from a patient safety perspective.
- Assign a provisional class (A, B, or C) to each requirement, based on the severity of harm that its failure could cause, using the definitions described above.
- Determine the overall software safety class. The classification of the entire software system is determined by the highest class assigned to any individual requirement.
Important considerations:
- Safety class assignments must be documented and justified with traceability to the risk management file (per ISO 14971).
- If a software item is composed of multiple software units, each unit can be individually classified — allowing lower-risk components to be developed under less stringent requirements.
- Classification should be reviewed and updated if requirements change during development.
Software Development Plan Required by IEC 62304
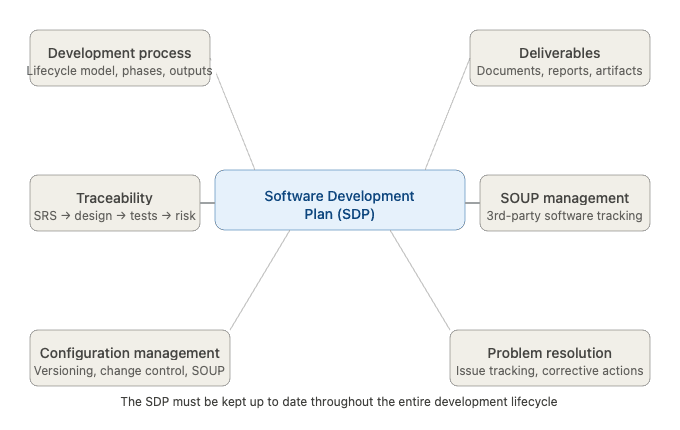
EC 62304 requires manufacturers to establish and maintain a Software Development Plan (SDP) — a living document that defines the framework for conducting all software lifecycle activities. This is not a bureaucratic formality: a well-structured SDP is a critical tool for ensuring quality, traceability, and regulatory compliance.
The Software Development Plan must address the following elements:
1. Development Process
Define the software development lifecycle model to be adopted (e.g., waterfall, iterative, agile-adapted) and describe how each phase will be executed, including activities, inputs, outputs, and acceptance criteria.
2. Deliverables
Identify all required outputs of each development activity — documents, code artifacts, test reports, review records — and specify who is responsible for producing and approving them.
3. Traceability
Establish and maintain bidirectional traceability between:
- Software requirements (SRS)
- Software architecture and design
- Implementation (code)
- Verification activities (tests)
- Risk control measures (per ISO 14971)
Traceability is essential for demonstrating that all requirements have been implemented and verified, and that all identified risks have been addressed.
4. Configuration and Change Management
Define how software versions, baselines, and changes are controlled throughout the lifecycle. This includes management of SOUP (Software of Unknown Provenance): third-party libraries, open-source components, and off-the-shelf software must be identified, versioned, and monitored for known anomalies.
5. Software Problem Resolution
Establish a process for identifying, evaluating, and resolving software-related problems — whether discovered during development, verification, or post-market surveillance. This feeds directly into the broader post-market monitoring obligations under MDR 2017/745 (in the EU context).
Critical note: The Software Development Plan must be kept up to date throughout the development process. If no update is made at a given stage, the rationale for not updating must be explicitly documented.
Software Architecture Under IEC 62304
IEC 62304 requires that a software architecture be defined and documented. The architecture must identify all software items (subsystems, components, units) and describe how they interact — including interfaces with hardware, external systems, and SOUP components.
Key requirements for the software architecture include:
- Partitioning of software items according to their safety class, ensuring that lower-class components cannot adversely affect higher-class ones without appropriate safeguards
- Identification of all interfaces between software items and between software and hardware
- Documentation of SOUP items used, including their version, manufacturer, and intended use within the system
- Ensuring the architecture supports the verification strategy — i.e., that individual units can be tested independently
The architecture document must be maintained in sync with the actual implementation and updated whenever significant architectural changes occur.
SOUP Management: An Overview According to IEC 62304
One topic that deserves specific attention within the IEC 62304 framework is the management of Software of Unknown Provenance (SOUP) — any software component not developed by the manufacturer but integrated into the medical device system. This includes open-source libraries, third-party modules, and commercial off-the-shelf (COTS) software.
IEC 62304 requires manufacturers to identify, document, and risk-assess all SOUP components used within the software system. Key obligations include maintaining a Software Bill of Materials (SBOM), performing integration testing, and establishing a change management process to monitor updates, security patches, and potential obsolescence throughout the product lifecycle.
From a cybersecurity standpoint, SOUP components are a critical attack surface: unmonitored third-party dependencies can introduce vulnerabilities that affect the safety and security of the entire device. This makes SOUP management not just a compliance checkbox, but an ongoing operational responsibility.
Given the complexity of this topic, a dedicated article on SOUP management under IEC 62304 is available — covering risk assessment, verification and validation, patch management, SBOM, and cybersecurity best practices in full detail.
AUDIT-READY KIT
Build your ISO 13485 QMS with confidence.
Built on 15+ years of audit experience — every SOP and template references the regulations auditors expect. Get to certification faster, with industry best practices baked in.
- ✓30 SOPs covering the full QMS scope
- ✓56 templates ready to customize
- ✓Aligned with EU MDR + FDA QMSR
Software Bill of Materials (SBOM): A Key Tool for SOUP Management
Closely linked to SOUP management is the concept of the Software Bill of Materials (SBOM) — a structured, comprehensive inventory of all software components present in a medical device system.
An SBOM provides visibility into software components (including third-party and open-source libraries), dependencies (nested software elements within other components), version numbers and licensing information, known vulnerabilities associated with included software, and sources and suppliers of each component.
Within the IEC 62304 framework, the SBOM serves as the practical implementation of several key requirements. While IEC 62304 does not explicitly mention the SBOM by name, its principles align with the concept by emphasizing software configuration management (Clause 8), software maintenance processes (Clause 6.4), and risk management of software components (Clause 4.3).
From a regulatory standpoint, the SBOM has also gained significant traction beyond IEC 62304. The FDA, in its Cybersecurity Guidance for Medical Devices, highlights the SBOM as a critical element in premarket submissions for cybersecurity risk management, requiring manufacturers to provide full transparency into their third-party and open-source software usage.
In practice, maintaining an up-to-date SBOM enables manufacturers to rapidly identify which components are affected when a new vulnerability is disclosed, streamline patch management decisions, and demonstrate compliance during regulatory audits — making it not just a documentation artifact, but an active risk management tool.
A dedicated article covering SBOM management in depth — including best practices, recommended formats (CycloneDX, SPDX), and integration with cybersecurity processes — is available for further reading.
Conclusions
IEC 62304 is far more than a regulatory checkbox — it is the foundational framework that enables medical device manufacturers to develop software that is safe, traceable, and fit for its intended clinical purpose. As this article has outlined, a correct understanding of software safety classification is the starting point for every subsequent decision: the rigor of the development process, the depth of verification activities, and the extent of SOUP and SBOM management all depend on whether a software system is classified as Class A, B, or C.
The Software Development Plan is the document that ties all of these activities together, providing a living record of how the organization intends to — and actually does — manage its software lifecycle. Combined with a well-defined software architecture and a disciplined approach to third-party components, it forms the backbone of a compliant and auditable development process.
It is worth emphasizing that IEC 62304 does not operate in isolation. In practice, it is always applied alongside ISO 14971 for risk management, IEC 62366 for usability engineering, and — increasingly — cybersecurity-specific standards such as IEC 81001-5-1. Together, these standards define the full regulatory picture for medical device software in markets including the EU (under MDR 2017/745) and the US (under FDA guidance).
As software continues to grow in complexity and connectivity — from embedded firmware to AI-based clinical decision support tools — mastering IEC 62304 becomes not just a compliance requirement, but a genuine competitive advantage. Manufacturers who build IEC 62304-compliant processes early in their development journey will find regulatory submissions smoother, post-market surveillance more manageable, and patient safety better protected.
This article is part of a series on medical device software regulation. The following articles belong to the same series:
- IEC 62304: The Complete Guide to the Medical Device Software Standard
- IEC 62304 Software Verification and Validation: Requirements by Safety Class
- IEC 81001-5-1: The Complete Guide to Cybersecurity for Health Software
- Medical Device Cybersecurity Testing: Methods, Requirements and Regulatory Compliance
- Software as a Medical Device (SaMD): Classification, Requirements and Regulatory Pathway